Isoseq_help_v1.0.html
说明文档
全长转录组简介
三代全长转录组测序和二代转录组测序的区别
大部分基因都不符合“一基因一转录本”的模式,基因往往存在多种剪切形式。通过二代测序,可以很准确地进行基因的表达及定量的研究,但是受限于读长的限制,不能得到全长转录本的信息。
基于二代测序平台的转录组产品,首先是把RNA打成小的短片断进行测序,然后再通过生物信息的方法进行拼接。但是基于二代测序平台的转录组,由于读长的限制(PE150),在转录本组装的过程中会存在较多的嵌合体,并且不能准确地得到完整转录本的信息,从而会大大降低表达量、可变剪接、基因融合等分析的准确性。下图是二代和三代转录组测序原理及读长比对:
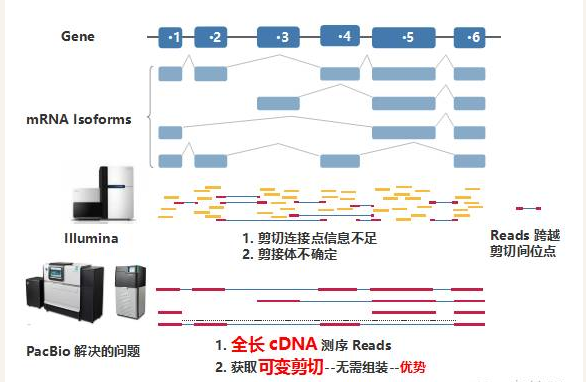
二代和三代转录组对比
| 分析内容 | 二代测序 | 三代测序 |
|---|
| 建库策略 | 一个文库 | 多级大片段文库 |
| 是否组装 | 是 | 否 |
| 差异表达基因 | 是 | 否 |
| 可变剪切 | 有局限 | 优 |
| 融合基因 | 有局限 | 优 |
| 正反义链 | 需要链特异性 | 直接测得 |
从上述可以看出,两种转录组测序技术互有优劣势,所以建议二+三代转录组测序技术同时使用,保证结构准确性、序列完整性及序列表达量准确性。
原始数据统计表
| 表头属性 | 说明 |
|---|
| Sample | 样品名称 |
| total base(bp) | 总碱基数 |
| subreads number | subreads数目 |
| average length | subreads平均长度 |
| N50 | subreads N50* |
* N50: 将所有subreads从大到小排序,并依次相加,当累加到所有subreads总长度的50%时, 该subread的长度即为所有subreads的N50。
CCS
Subread:指原始序列中的每个子序列,不包括adapter序列。如下示意图。
CCS(Circular Consensus Sequence):指原始序列中的所有subreads校正后得到的cDNA序列。
Full passes:一个Full passes指原始序列中的一条subread两端均含有 Adapter,一个原始序列的Full passes数目指在该序列中文库cDNA序列被完整测到的次数。
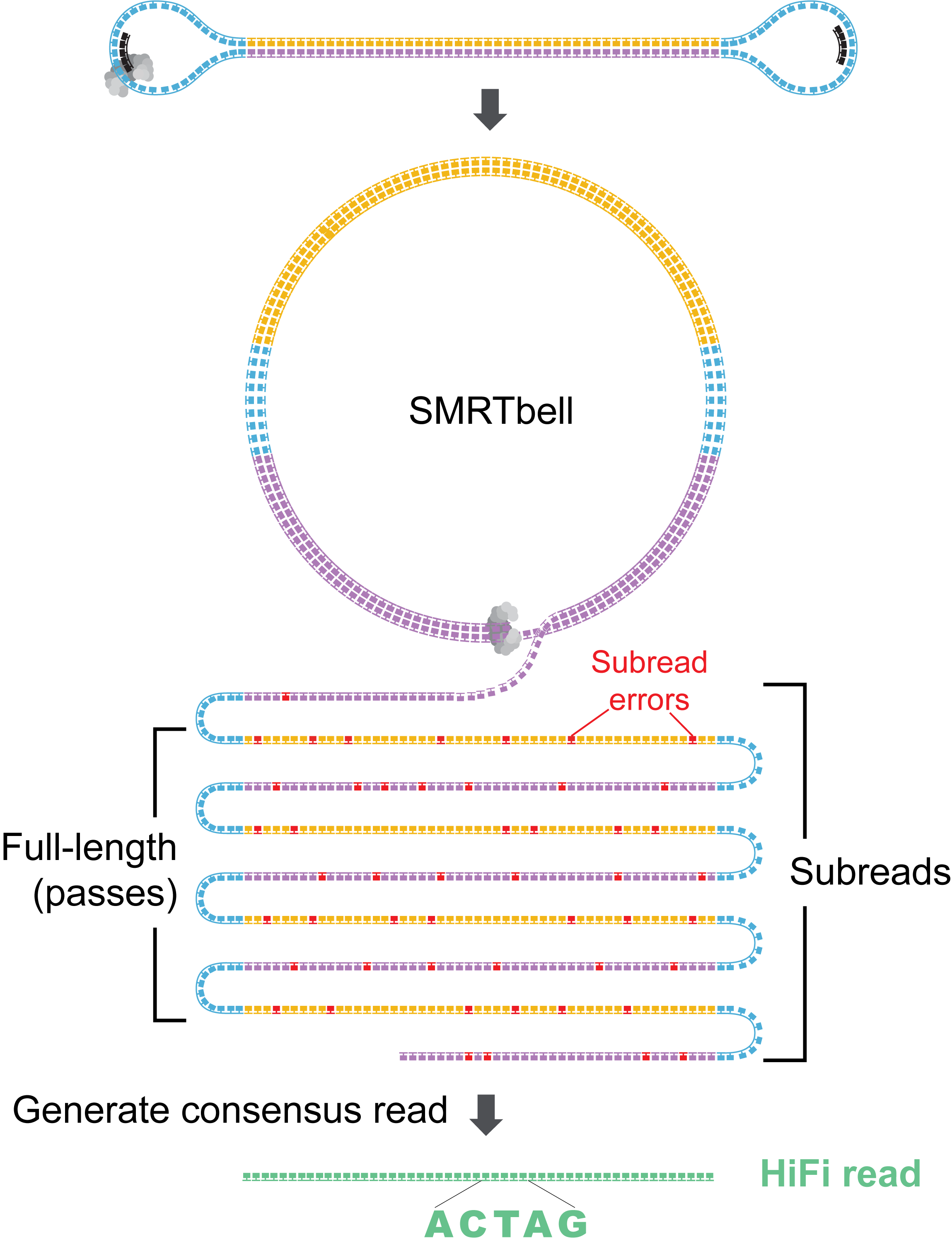
图3-2 Subreads, CCS示意图
CCS分析结果表
| 表头属性 | 说明 |
|---|
| Sample | 样品名称 |
| Number of reads | CCS reads数量 |
| Number of CCS bases | CCS碱基数量 |
| CCS Read Length (mean) | CCS Read平均长度 |
| Number of Passes (mean) | 平均full Pass数量 |
获取FLNC
FL:full-length reads,全长序列。指包含了5’ primer, 3’ primer和poly(A)的ccs序列。
FLNC:full-length non-contatemer,全长非嵌合序列,是指去除了多联体的全长序列
多联体:是指一条reads中包含了两条或以上转录本序列,可能在文库制备阶段或PCR反应中产生。
- Artificial Concatemer

- PCR Chimera

Classify分析结果表
| 表头属性 | 说明 |
| Sample | 样品名称 |
| CCS reads number | 过滤后的CCS序列数量 |
| FLNC reads number | FLNC序列数量 |
| Percentage of FLNC in CCS | FLNC占CCS序列百分比 |
| Mean length of FLNC | FLNC平均长度 |
Reads聚类和校正
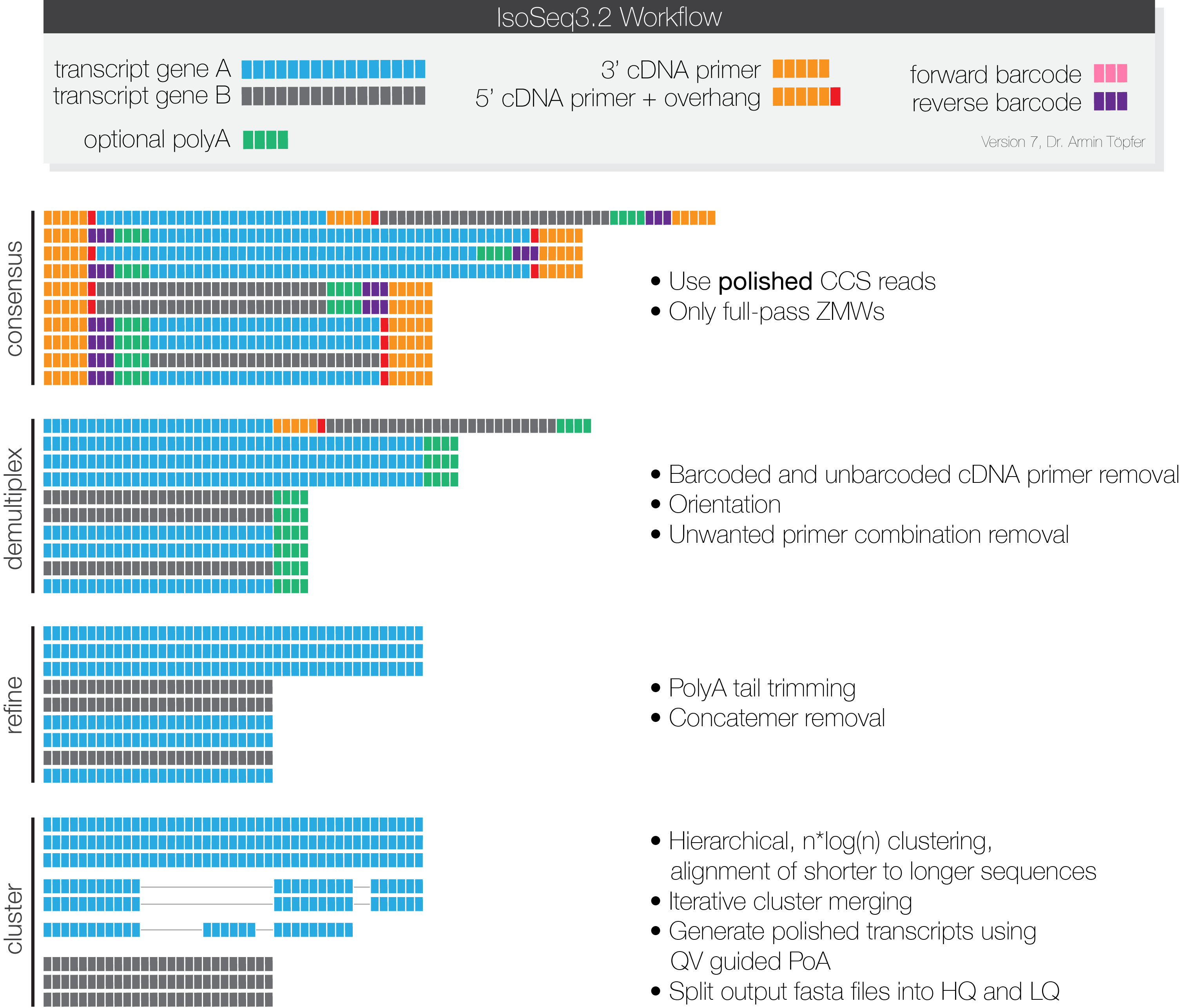
图3-5 Reads聚类与校正示意图
Cluster分析结果表
| 表头属性 | 说明 |
|---|
| Number of polished high-quality isoforms | 经校正的高质量序列数量 |
| Number of polished low-quality isoforms | 经校正的低质量序列数量 |
* 高质量序列是指准确度大于99%的序列
转录本的校正
使用LoRDEC软件进行转录本校正,LoRDEC采用混合策略,需要使用两组数据:参考reads(二代短reads)、PacBio长reads。通过读取短reads,建立de Bruijn图,针对长reads中的错误区域寻找校正序列,详细介绍请参考:http://www.atgc-montpellier.fr/lordec/
| Total number | 校正后序列数量 |
| Total length (bp) | 总碱基数 |
| Maximum Length (bp) | 最长序列碱基数 |
| Minimum Length (bp) | 最短序列碱基数 |
| Average Length (bp) | 序列平均碱基数 |
| N50 Length | N50的长度 |
| GC content | GC含量 |
序列去冗余
去冗余之后isoform统计表说明:
| Total number | isoform序列数目 |
| Total length (bp) | isoform序列总长度 |
| Maximum Length (bp) | 最长isoform的长度 |
| Minimum Length (bp) | 最短isoform的长度 |
| Average Length (bp) | isoform平均长度 |
| N50 Length | N50的长度 |
| GC content | GC含量 |
基本注释
四大数据库注释统计表:
| Total Isoform | 全长转录本总数 |
| Nr | 能注释到Nr数据库的全长转录本数 |
| Swissport | 能注释到Swissport数据库的全长转录本数 |
| KOG | 能注释到KOGt数据库的全长转录本数 |
| Kegg | 能注释到Kegg数据库的全长转录本数 |
| annotation gene | 有注释的全长转录本的数目 |
| without annotation gene number | 没有注释的全长转录本总数 |
注释结果汇总表各列含义如下所示:
| geneID | Gossypium_australe-Isoform序列的 ID 号 |
| Symbol | Symbol 号 |
| Nr-ID | blast 比对到 Nr 数据库中序列名 |
| Nr-Score | blast 比对到 Nr 数据库的 Score |
| Nr-Evalue | blast 比对到 Nr 数据库的 Evalue |
| Nr-annotation | Nr 序列的描述 |
| Swissprot-ID | blast 比对到 Swissprot 数据库中序列名 |
| Swissprot-Score | blast 比对到 Swissprot 数据库的 Score |
| Swissprot-Evalue | blast 比对到 Swissprot 数据库的 Evalue |
| Swissprot-annotation | Swissprot 序列的描述 |
| COG/KOG-Protein-or-Domain | blast 比对到 COG/KOG 数据库中的蛋白或结构域 |
| COG/KOG-Score | blast 比对到 COG/KOG 数据库的 Score |
| COG/KOG-Evalue | blast 比对到 COG/KOG 数据库的 Evalue |
| COG/KOG-ID | COG/KOG 的 ID |
| COG/KOG-Function-Description | COG/KOG 功能描述 |
| KO-ID | KO 的 ID及注释 |
| KEGG-Evalue | blast 比对到 KEGG 数据库的 Evalue |
| KEGG-Score | blast 比对到 KEGG 数据库的 Score |
| KEGG-Gene | blast 比对到 KEGG 数据库中的序列名 |
| Pathway | Pathway |
| GO-BiologicalProcess | GO生物过程 |
| GO-MolecularFunction | GO分子功能 |
| Go-CellularComponent | GO细胞组分 |
blast比对各个数据库结果表各列含义如下所示:
| Query_id | Isoform序列的 ID 号 |
| Subject_id | 比对到数据库中序列名 |
| Identity | 比对的identity |
| Align_length | 比对上的长度 |
| Miss_match | 比对的错配数 |
| Gap | Gap的比例 |
| Query_start | 比对上的部分在 Isoform上的起始位置 |
| Query_end | 比对上的部分在 Isoform上的终止位置 |
| Subject_start | 比对上的部分在数据库序列上的起始位置 |
| Subject_end | 比对上的部分在数据库序列上的终止位置 |
| E_value | 比对的Evalue |
| Score | 比对的分值 |
| Subject_annotation | 数据库序列的描述 |
物种分布统计表各列含义如下所示:
| Species | 物种名称 |
| Isoform Num | 与物种同源的 Isoform 数目 |
COG/KOG
“COG/KOG”是 Cluster of Orthologous Groups of proteins(蛋白相邻类的聚簇)的缩写。构成每个 COG/KOG 的蛋白都是被假定为来自于一个祖先蛋白,并且因此或者是 orthologs 或者是 paralogs
Orthologs 是指来自于不同物种的由垂直家系(物种形成)进化而来的蛋白,并且典型的保留与原始蛋白有相同的功能
Paralogs是那些在一定物种中的来源于基因复制的蛋白,可能会进化出新的与原来有关的功能
KOG/COG 分类表表各列含义如下:
| Code | COG/KOG 功能代号 |
| Functional-Categories | COG/KOG 功能分类 |
| Gene-Number | Isoform在各 COG/KOG 功能分类的数量 |
| Genes | Isoform在各 COG/KOG 功能分类的序列ID |
KOG/COG注释表表各列含义如下:
| Gene | Isoform序列的 ID 号 |
| Protein-or-Domain | 比对上的蛋白或结构域 |
| E-Value | blast 比对的 Evalue |
| COG/KOG-ID | COG/KOG 的 ID |
| Function-Description | 功能描述 |
| Code | COG/KOG 功能代号 |
| Functional-Categories | COG/KOG 功能分类 |
KEGG
KEGG(京都基因与基因组百科全书)是基因组破译方面的数据库。在后基因时代一个重大挑战是如何使细胞和有机体在计算机上完整的表达和演绎,让计算机利用基因信息对更高层次和更复杂细胞活动和生物体行为作出计算推测。为达到此目的,人们建立了一个在相关知识基础上的网络推测计算工具。在给出染色体中一套完整的基因的情况下,它可以对蛋白质交互(互动)网络在各种细胞活动起的作用作出预测。 KEGG 的 PATHWAY 数据库整合当前在分子互动网络(比如通道,联合体)的知识,KEGG 的 GENES/SSDB/KO 数据库提供关于在基因组计划中发现的基因和蛋白质的相关知识,KEGG 的 COMPOUND/GLYCAN/REACTION 数据库提供生化复合物及反应方面的知识。
KEGG 是系统分析基因产物在细胞中的代谢途径以及这些基因产物的功能的数据库,利用 KEGG 可以进一步研究基因在生物学上的复杂行为。根据 KEGG 注释信息我们能进一步得到 Unigene的 Pathway 注释。
KEGG 注释表各列含义如下:
| Pathway | 通路名 |
| Count (*) | 注释到该通路的基因的数目 |
| Pathway ID | KEGG 数据库中的 Pathway ID |
| Genes | 注释到该 Pathway 的基因 |
| KOs | 属于该 Pathway 的 KEGG Orthology |
GO
根据 nr 注释信息我们能得到 GO 功能注释。Gene Ontology(简称GO)是一个国际标准化的基因功能分类体系,提供了一套动态更新的标准词汇表(controlled vocabulary)来全面描述生物体中基因和基因产物的属性。
GO 总共有三个 ontology,分别描述基因的分子功能(molecular function)、细胞组分(cellular component)、参与的生物过程(biological process)。
GO 的基本单位是 term(词条、节点),每个 term 都对应一个属性。GO 功能分析一方面给出差异表达基因的 GO 功能分类注释;另一方面给出差异表达基因的 GO 功能显著性富集分析。
我们根据 nr 注释信息,使用 Blast2GO软件得到 Isoform的 GO 注释信息。Blast2GO 已被其它文献引用超过 150 次,是同行广泛认可的 GO 注释软件。
得到每个 Isoform的 GO 注释后,我们用 WEGO 软件对所有 Isoform做 GO 功能分类统计,从宏观上认识该物种的基因功能分布特征。
GO 注释表各列含义如下:
geneID 序列的 ID 号
GO GO ID
GO 分类表各列含义如下:
| Ontology | GO本体的类别 (biological_process 或 cellular_component 或 molecular_function) |
| Class | GO 条目 |
| number_of_* | Isoform在注释到各 GO 条目 中的数量 |
| genes_of_* | 注释到各 GO 条目的 IsoformID |
GO分类图示例
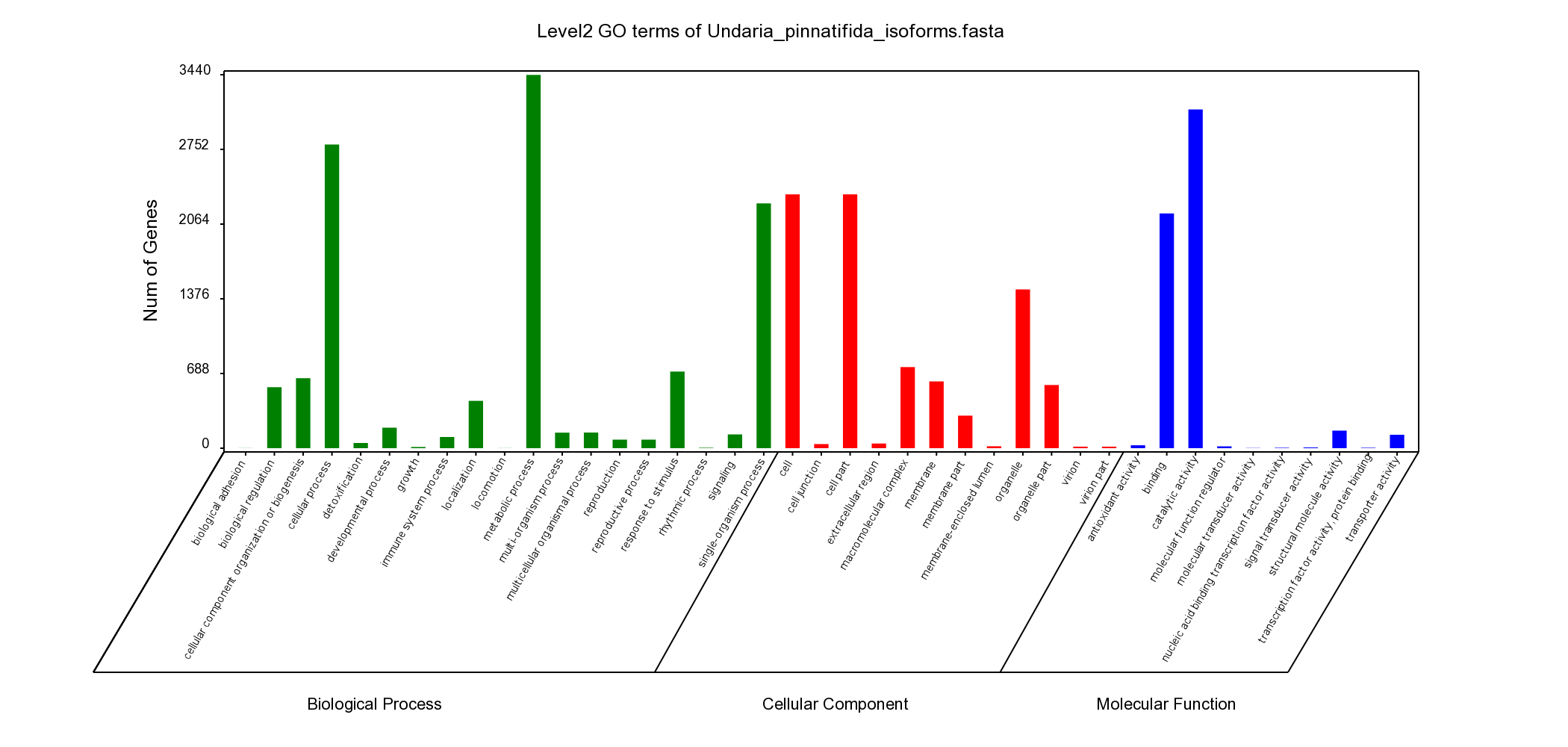
该图根据 GO分类表对 Isoform的 GO 注释进行统计,横坐标代表 GO 三个 ontology:分子功能(molecular function)、细胞组分(cellular component)、参与的生物过程(biological process)的更细一级分类;纵坐标代表每个分类条目所对应的基因数目。由于一个基因常常会有多个不同功能,因此同一个基因会在不同分类条目下出现,每个柱状图统计相互独立。
CDS 预测
CDS预测汇总表格说明
| ID | isoform ID |
| 5UTR start | 5'UTR 的开始位点 |
| 5UTR end | 5’UTR 的结束位点 |
| CDS start | CDS 的开始位点 |
| CDS end | CDS 的结束位点 |
| 3UTR start | 3'UTR 的开始位点 |
| 3UTR end | 3'UTR 的结束位点 |
| Symbol | Symbol 号 |
| Nr-ID | blast 比对到 Nr 数据库中序列名 |
| Nr-Score | blast 比对到 Nr 数据库的 Score |
| Nr-Evalue | blast 比对到 Nr 数据库的 Evalue |
| Nr-annotation | Nr 序列的描述 |
| Swissprot-ID | blast 比对到 Swissprot 数据库中序列名 |
| Swissprot-Score | blast 比对到 Swissprot 数据库的 Score |
| Swissprot-Evalue | blast 比对到 Swissprot 数据库的 Evalue |
| Swissprot-annotation | Swissprot 序列的描述 |
| COG/KOG-Protein-or-Domain | blast 比对到 COG/KOG 数据库中的蛋白或结构域 |
| COG/KOG-Score | blast 比对到 COG/KOG 数据库的 Score |
| COG/KOG-Evalue | blast 比对到 COG/KOG 数据库的 Evalue |
| COG/KOG-ID | COG/KOG 的 ID |
| COG/KOG-Function-Description | COG/KOG 功能描述 |
| KO-ID | KO 的 ID及注释 |
| KEGG-Evalue | blast 比对到 KEGG 数据库的 Evalue |
| KEGG-Score | blast 比对到 KEGG 数据库的 Score |
| KEGG-Gene | blast 比对到 KEGG 数据库中的序列名 |
| Pathway | Pathway |
| GO-BiologicalProcess | GO生物过程 |
| GO-MolecularFunction | GO分子功能 |
| Go-CellularComponent | GO细胞组分 |
可变剪切分析
Cogent
Cogent (COding GENome reconstruction Tool), 是一款在无参考基因组的情况下,寻找基因家族并重构编码基因序列的工具。具体方法如下图所示:
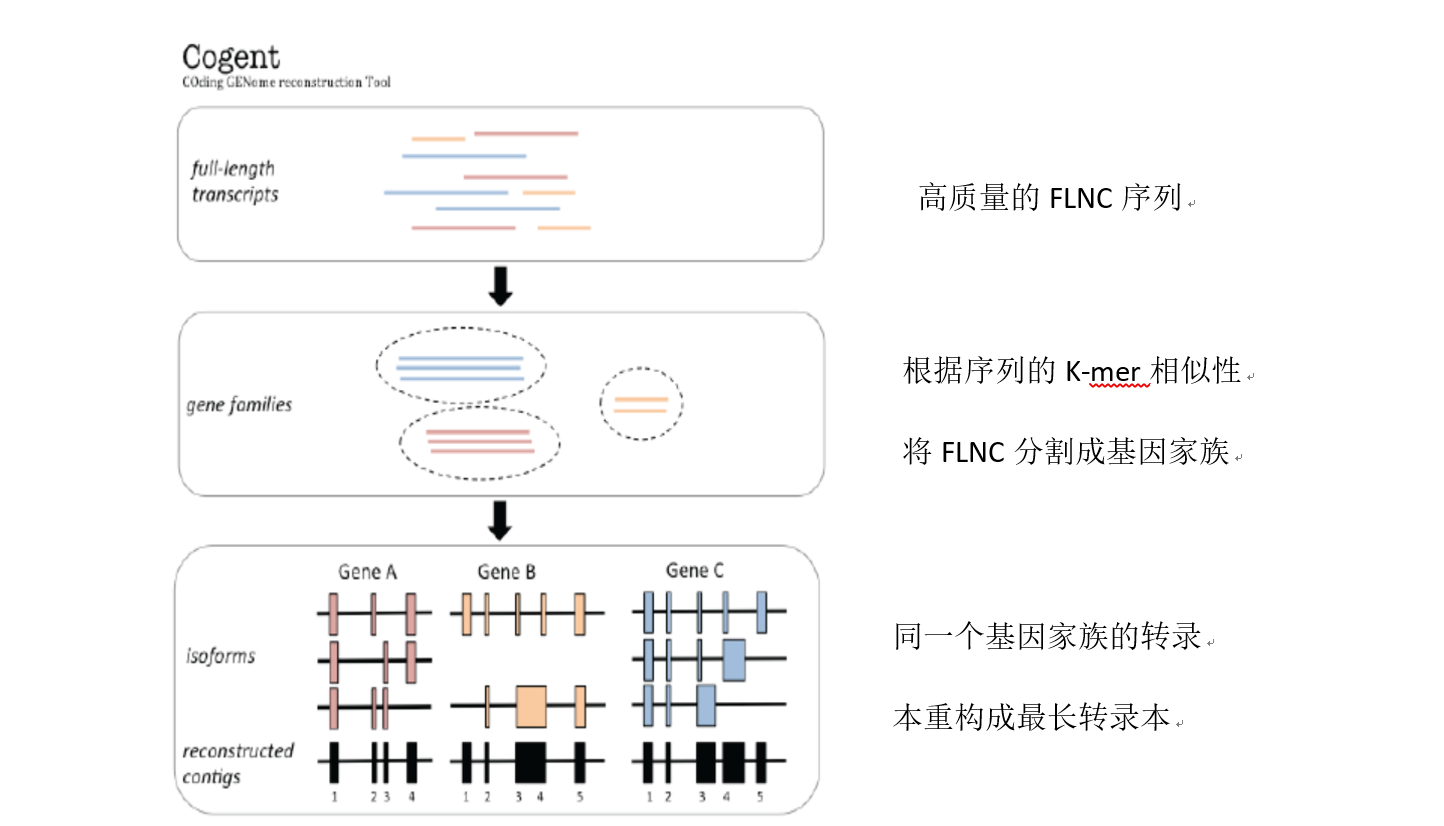
第一步:Cogent软件读取高质量的full-length non- chimeric (FLNC)序列
第二步:Cogent软件根据FLNC序列的K-mer相似性(K-mer=30,相似度=0.95,K-mer similarity>95%)将FLNC序列分割成基因家族,每个基因家族包含一条或多条FLNC序列
第三步:同一个基因家族的FLNC序列构建最长最全转录本。
例如:一个基因家族中有3条FLNC序列,第一条有1,2,3号外显子,第二条有1,3,4号外显子,第三条有1,4号外显子,那么重构的这个基因家族的的最长最全转录本包含有1,2,3,4四个外显子,原本的三条FLNC序列去掉,保留最长最全的这个转录本作为参考序列。
组装出参考序列后,使用软件SUPPA进行可变剪切分析。SUPPA是一款能够进行可变剪切事件分类和差异可变剪切表达分析的软件。具体介绍请参考:https://github.com/comprna/SUPPA
可变剪切表格解析
SUPPA对可变剪切事件分为7类::
- SE: Skipping Exon
- A5: Alternative 5' Splice Site
- A3: Alternative 3' Splice Site
- MX: Mutually Exclusive Exon
- RI: Retained Intron
- AF: Alternative First Exon
- AL: Alternative Last Exon
可变剪切位点信息解析:
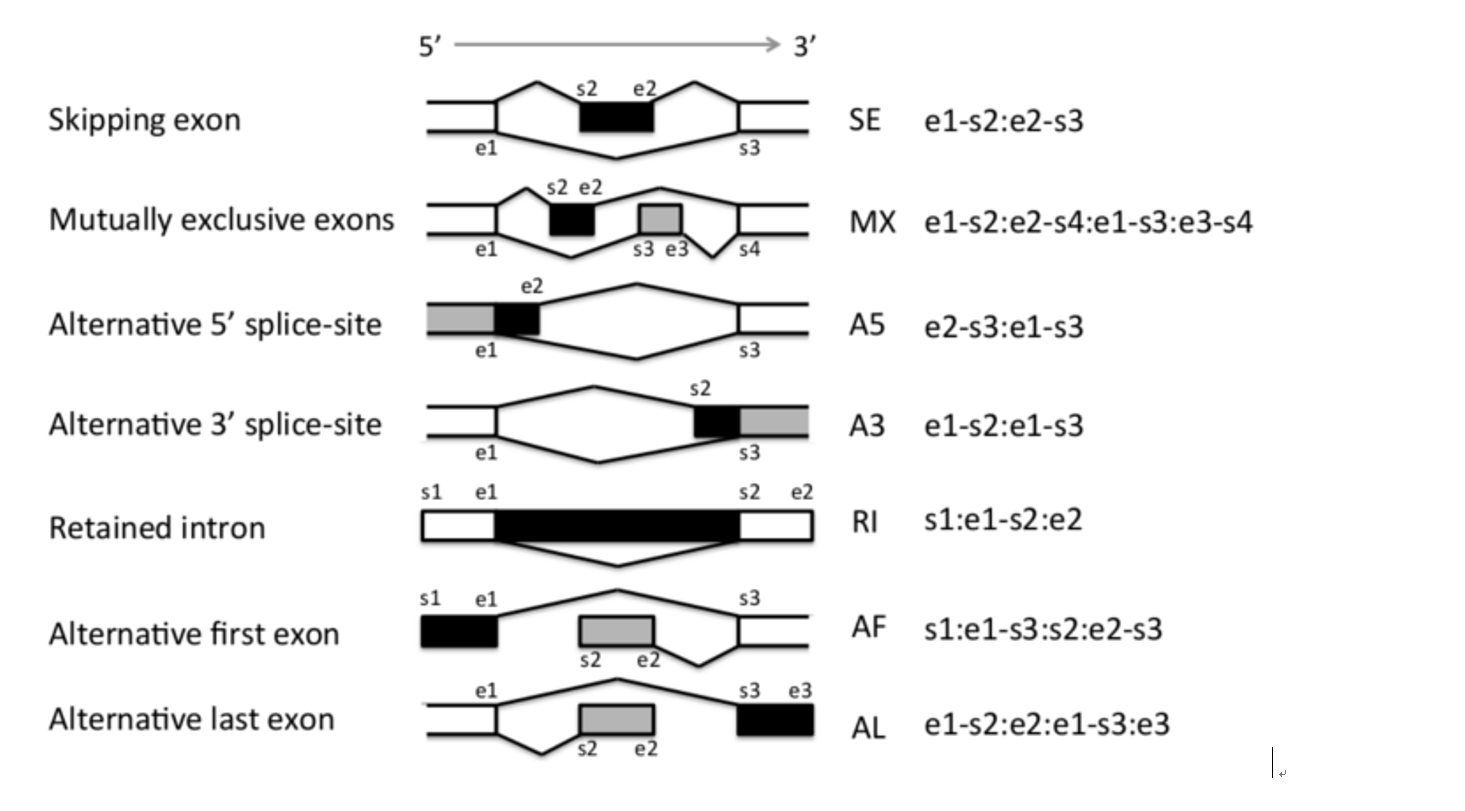
可变剪切表格解析
| seqname | Cogent 组装出来的序列ID |
| Type | 可变剪切类型 |
| pos | 可变剪切位点信息 |
| Strand | 正负链 |
| total_transcripts | 发生可变剪切的全部转录本 |
串联重复单元(SSR)
我们按照以下配置参数使用软件 MISA(http://pgrc.ipk-gatersleben.de/misa/)对转录组的所有 Isoform进行搜索,寻找 Isoform中的 SSR :
配置参数信息:
definition ( unit_size,min_repeats ) : 2-6 3-5 4-4 5-4 6-4
interruptions ( max_difference_between_2_SSRs ) : 100
备注:
1)(n,m): n,串联重复单元的长度;m,最小重复数;例如:2-6说明对于2个核苷酸的重复单元,需要至少6个重复才会被认为是SSR
2)interruptions:如果两个SSR序列的距离短于100bp,就会被合并当做一个SSR标记
SSR引物设计汇总表各列含义:
| ID | Isoform编号 |
| SSR nr. | 对于 Isoform的 SSR 序号,如 2,表示该 SSR 是此 Isoform上的第 2 个 SSR |
| SSR type | SSR 类型,如 P3,表示重复拷贝数为 3,c 表示该 SSR 标记序列中的短串联重复序列不止一条 |
| SSR | 短串联重复序列 |
| size | 序列的长度 |
| start | SSR 序列在 Isoform上的起始位置 |
| end | SSR 序列在 Isoform上的终止位置 |
| FORWARD PRIMER1 (5'-3') | 正向引物 1 |
| Tm (°C) | 正向引物的 Tm 值 |
| size | 正向引物的长度 |
| REVERSE PRIMER1 (5'-3') | 反向引物 1 |
| Tm (°C) | 反向引物的 Tm 值 |
| size | 反向引物的长度 |
| PRODUCT1 size (bp) | 引物扩增产物的长度 |
| ...... | 另外两对引物的设计结果 |
结构域分析(Pfam)
Pfam(Protein families database of alignments and hidden Markov models )是一个基于多重序列比对以及隐马尔可夫模型(HMM)预测的方法而收录的大量蛋白质结构信息的数据库,被广泛用来做蛋白结构域预测及蛋白家族分析。
Pfam 包括 PfamA 和 PfamB。其中 PfamA 中所包含的蛋白结构数据都是已知并且得到验证的,每个蛋白结构域都有各自的定义(definition)。而 PfamB 中的数据是通过模型和算法预测出来的,并且未得到验证,是对 PfamA 的补充。
Pfam 蛋白结构相关注释汇总表各列含义:
| seq id | Isoform的编号 |
| alignment start | Isoform编码蛋白序列比对的结构域起始位置 |
| alignment end | Isoform编码蛋白序列比对的结构域终止位置 |
| envelope start | HMM 模型预测的 Isoform编码蛋白序列的结构域起始位置 |
| envelope end | HMM 模型预测的 Isoform编码蛋白序列的结构域终止位置 |
| hmm acc | Isoform编码蛋白序列对应结构的 HMM 在 Pfam 中的编号 |
| hmm name | Isoform编码蛋白序列对应结构的 HMM 在 Pfam 中的名称 |
| type | Isoform编码蛋白序列匹配到 Pfam 数据库中对应结构的分类水平,蛋白家族或者结构域 |
| hmm start | 数据库中匹配序列的起始位置 |
| hmm end | 数据库中匹配序列的终止位置 |
| hmm length | 数据库中匹配序列的长度 |
| bit score | 根据比对和 HMM 模型得出的 Isoform编码蛋白序列结构的评分 |
| E-value | 比对注释的假阳性率 |
| significance | Isoform编码蛋白序列在数据库中匹配结构的数目 |
| clan | Pfam 数据库中按照蛋白质序列,结构以及 HMM 文件而分成的类群 |
| PfamA_definition | 查询序列对应结构在 PfamA 中的名称 |
结构域分析(SMART)
SMART 蛋白结构相关注释汇总表各列含义:
| GeneID | Isoform的编号 |
| Length | Isoform的长度 |
| Domain_Start | Isoform上该 SMART Domain 的起始位置 |
| Domain_End | Isoform上该 SMART Domain 的终止位置 |
| SMART_Domain | SMART Domain 名称 |
| E-value | Sequence E-value |
| Score | Sequence Bit Score |
| Domain-c-Evalue | Domain Conditional E-value |
| Domain-i-Evalue | Domain Independent E-value |
| Domain-Score | Domain Bit Score |
TF 转录因子分析
TF 相关注释汇总表各列含义:
| gene_id | Isoform的编号 |
| TF_Family | TF 家族 |
R-Gene 分析
Rgene 相关注释汇总表各列含义:
| Unigene_ID | | Isoform的编号 |
| PRGID | | PRG 数据库编号 |
| Name | | R-Gene 名称 |
| Type | | R-Gene 类型 |
| Species | | R-Gene 来源物种 |
| Class | | R-Gene 分类 |
| GenBank ID | | GenBank ID |
| GenBank Locus | | GenBank Locus |
| Description | | 功能描述 |
| E-value | | E-value |
| Score | | Bit Score |
lncRNA 分析
lncRNA结果统计表说明:
| Sample | | 样本名 |
| total isoform | | isoform 总数 |
| the number of mRNA | | mRNA的数目 |
| the number of lncRNA | | 预测到的lncRNA数目 |
lncRNA家族预测结果表格说明:
| Family_Name | | 在Rfam数据库中家族的名字 |
| Family_Accession | | 家族的编号 |
| LncRNA | | lncRNA的ID |
| Strand | | 正负链 |
| E-value | | 比对的的E值 |
| Score | | Infernal给出的比对的分值 |
蛋白相关分析补充
|
|
|
| Unigene_ID | Unigene的编号 |
| Length | 该Unigene编码的蛋白氨基酸序列的长度 |
| ExpAA | 预测的跨膜螺旋氨基酸数目。如果这个数值大于18,则该蛋白很可能是一个跨膜蛋白,或者含有信号肽结构 |
| First60 | 蛋白N端前60个氨基酸中预测为跨膜螺旋氨基酸的数目。预测N端的跨膜螺旋结构为信号肽的可能性 |
| PredHel | 通过N-best预测的跨膜螺旋数量 |
| Topology | 由N-best拓扑预测的最终跨膜结果。"i"表示预测该蛋白在膜内;"o"表示预测该蛋白在膜外;"i95-117o165-187i"表示从1-94号氨基酸肽链在膜内,95-117号氨基酸肽链为跨膜螺旋结构,118-164号氨基酸肽链在膜外,165-187号氨基酸肽链为跨膜结构 |
| Unigene_ID | | Unigene的编号 |
| Cleavage_Site | | 预测的信号肽剪切位点 |
| Cmax | | 最大C值。C值:剪切位点得分。每个氨基酸都会有一个C值,在剪切位点处C值最高 |
| C-pos | | 最大C值位置 |
| Ymax | | 最大Y值。Y值:综合考虑C值和S值的剪切位点得分,比单独考虑C值要更精确 |
| Y-pos | | 最大Y值位置 |
| Smax | | 最大S值。S值:信号肽得分。每个氨基酸对应一个S值,信号肽区域的S值较高 |
| S-pos | | 最大S值位置 |
| Smean | | 从N端氨基酸开始到剪切位点处各氨基酸的平均S值 |
| D | | S-mean和Y-max的平均值,对区分是否为分泌蛋白具有重要作用 |
| If_SignalP | | 是否预测为分泌蛋白 |
NetOGlyc 结果表各列含义:
| Unigene_ID | Unigene的编号 |
| Length | 该Unigene编码的蛋白氨基酸序列的长度 |
| Oglyc_Site | O-型糖基化位点 |
| S_num | 序列中Ser丝氨酸数目 |
| T_num | 序列中Thr苏氨酸数目 |
| S_and_T_num | 序列中S+T数目 |
| Oglyc_Site_num | 序列中O-型糖基化位点数目 |
ProP 结果表各列含义:
| Unigene_ID | Unigene的编号 |
| Length | 该Unigene编码的蛋白氨基酸序列的长度 |
| Prop_Site | 弗林蛋白酶裂解位点 |
| R_num | 序列中Arg精氨酸数目 |
| K_num | 序列中Lys赖氨酸数目 |
| R_and_K_num | 序列中R+K数目 |
| Prop_Site_num | 序列中弗林蛋白酶裂解位点数目 |
|