1 项目概述
| 项目编号 | GDMR21010245_sup_6 |
| 样品信息 | LL-1¦LL-2¦LL-3¦LN-1¦LN-2¦LN-3
TL-1¦TL-2¦TL-3¦TN-1¦TN-2¦TN-3 |
| 分组方案 | LL :LL-1&LL-2&LL-3¦LN :LN-1&LN-2&LN-3¦TL :TL-1&TL-2&TL-3¦TN :TN-1&TN-2&TN-3 |
| 两组差异分析方案 | LN-vs-LL¦TN-vs-TL¦LL-vs-TL¦LN-vs-TN |
2 项目简介
2.1 背景介绍
由转录组和代谢组数据获得了样本在基因表达水平和代谢物水平的差异特征。然而生物系统中转录与代谢并不是独立发生的,为揭示基因表达与代谢物之间的调控影响机制,可基于“参与同一生物过程的基因或代谢物具有相同或相似的变化规律”进行二者的关联分析。
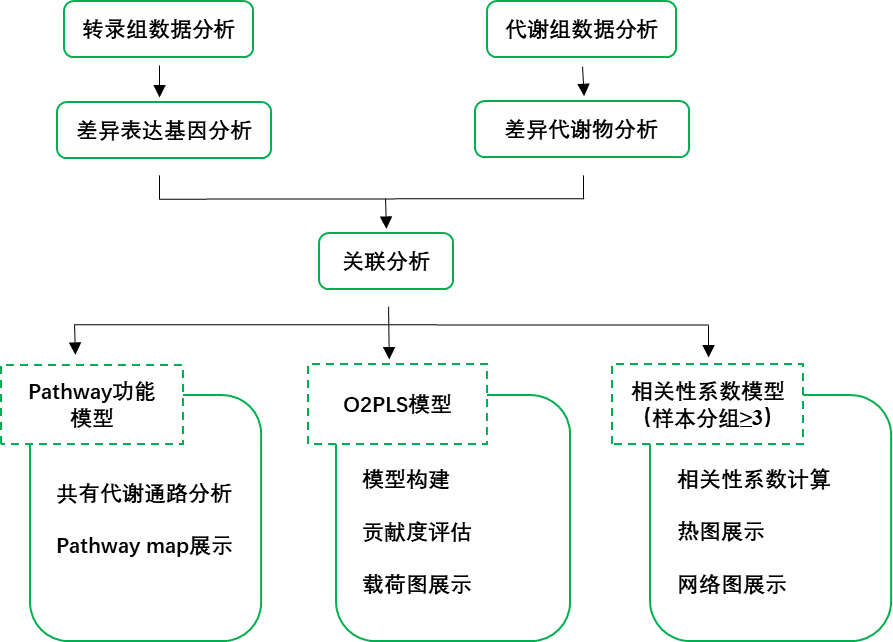 |
为筛选获得对样本分组有影响的关联基因与代谢物集合,分析关联特征,基于基因表达量和代谢物丰度两组数据进行3种模型分析:1)Pathway功能模型,查询基因与代谢物共有的KEGG 代谢通路(pathway),分析共有pathway中基因与代谢物的关联特征;2)O2PLS(Bidirectional orthogonal projections to latent structures)模型,使用基因表达量与代谢物丰度数据构建 O2PLS模型,通过模型预测,获得有关联性的基因与代谢物集合并分析;3)相关性系数模型(当样本分组 ≥ 3时,进行此模型分析),计算基因表达量与代谢物丰度的pearson相关性系数,并以热图和网络图展示。 |
| Fig 2-1-1 信息分析流程图 |
3 Pathway功能模型
3.1 共有代谢通路分析
通过组间差异分析,由转录组数据获得了差异表达的基因,由代谢组数据获得了差异表达的代谢物,并进行了各自组学的KEGG富集分析。在关联分析中,将针对基因与代谢物进行共有KEGG pathway的分析[1][2][3]。包含三种类型:
1)组间差异基因与组间差异代谢物共有pathway的分析;
2)由于组间差异代谢物的种类可能很少,或者差异代谢物中没有目标代谢物,故进行组间差异基因与所有代谢物共有pathway的分析;
3)作为其他个性化分析筛选的基础,进行所有基因与所有代谢物共有pathway的分析。
| 关联类型 | 基因数据来源 | 代谢物数据来源 | 共有pathway | Pathway 注释表 |
|---|---|---|---|---|
| 差异基因与差异代谢物 | LN-vs-LL | LN-vs-LL | LN-vs-LL.htm | LN-vs-LL.path.xls |
| TN-vs-TL | TN-vs-TL | TN-vs-TL.htm | TN-vs-TL.path.xls | |
| LL-vs-TL | LL-vs-TL | LL-vs-TL.htm | LL-vs-TL.path.xls | |
| LN-vs-TN | LN-vs-TN | LN-vs-TN.htm | LN-vs-TN.path.xls | |
| 差异基因与所有代谢物 | LN-vs-LL | all | LN-vs-LL_all.htm | LN-vs-LL_all.path.xls |
| TN-vs-TL | all | TN-vs-TL_all.htm | TN-vs-TL_all.path.xls | |
| LL-vs-TL | all | LL-vs-TL_all.htm | LL-vs-TL_all.path.xls | |
| LN-vs-TN | all | LN-vs-TN_all.htm | LN-vs-TN_all.path.xls | |
| 所有基因与所有代谢物 | all | all | all.htm | all.path.xls |
3.2 Pathway map
点击共有pathway分析页面的KEGG描述,可链接基因与代谢物相关联的pathway map,示例如下:
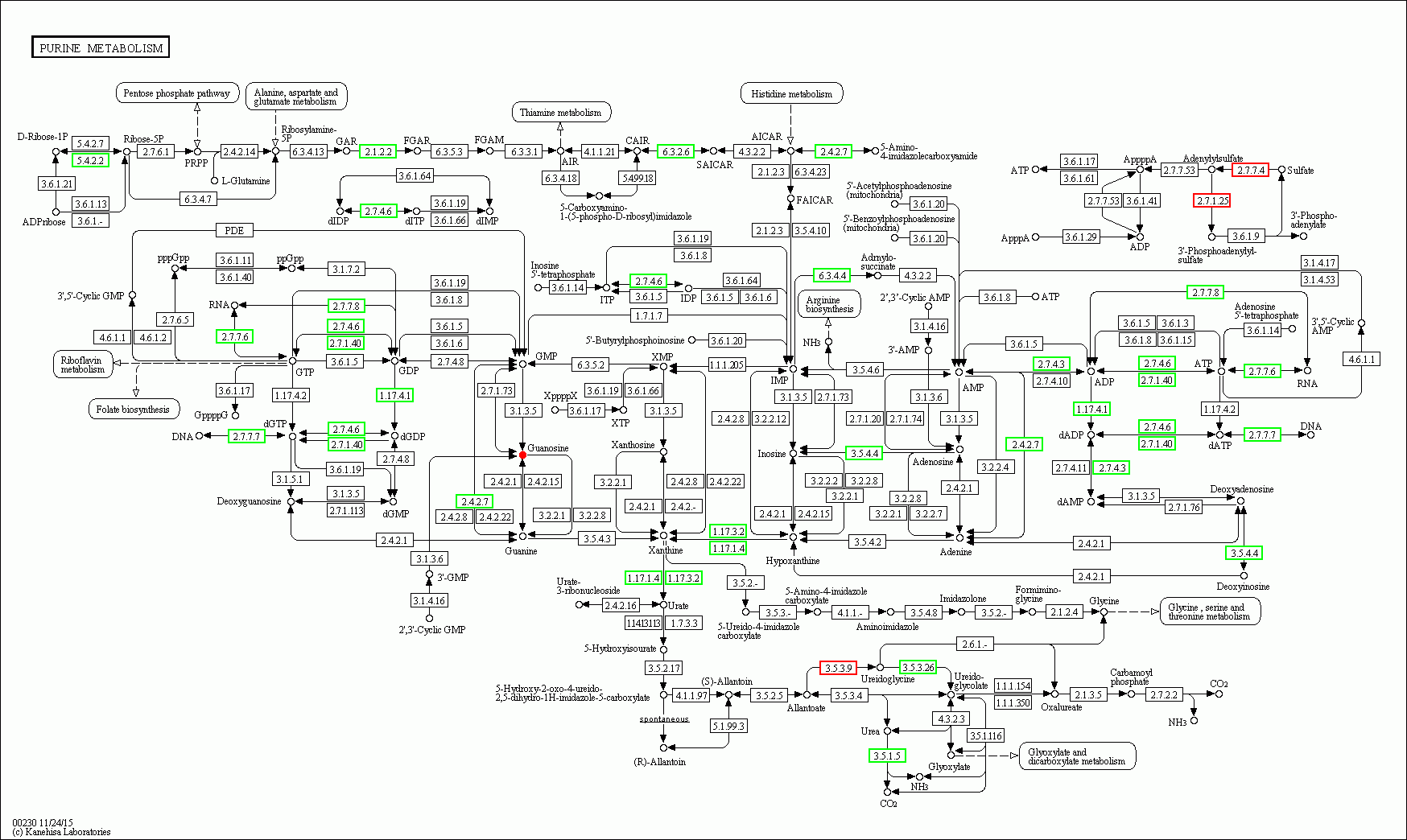 |
| Fig 3-2-1 差异代谢物与差异基因关联示例 |
4 O2PLS模型分析
基于所有转录组和所有代谢组数据,我们利用OmicsPLS包开展O2PLS分析[4]。O2PLS模型适用于两个组学数据的关联分析,可在两个数据矩阵中进行双向建模和预测。O2PLS模型通过计算,将每个组学的数据都分解为三个部分,即关联部分(joint part,即与另一组学关联程度很大的部分)、正交部分(Systematic/Orthogonal part,即只对本组学数据有影响、而对另一组学数据没有影响的部分)、噪音部分(noise part,即对两组学数据都没有影响的部分),如下图所示。各部分对总变异的解释程度以R2表示,值越高表示模型的解释能力越好。模型的过拟合和不足都会降低R2。
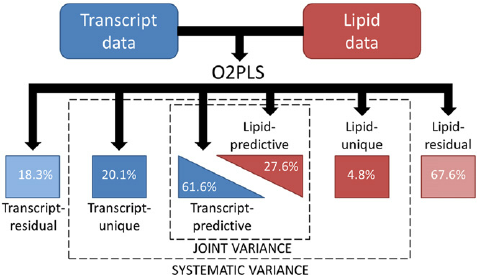 |
| Fig 4-0-1 o2pls模型示例 |
OmicPLS包进行O2PLS模型分析的步骤如下:
1)对两个独立数据集(X,Y,如基因表达量和代谢物丰度数据)分别进行标准化,使总平方和为1,以消除组学数据数量级差异的影响,使统计分析更科学。
2)Cross-validation:确定组分个数。以关联部分组分个数a大于1,基因正交部分组分个数ax和代谢组正交部分组分个数ay大于0开始,进行多次不同数值组合的预建模,计算预测误差。
3)Fitting:最适建模。选取预测误差最小时,对应的组分个数n,nx,ny来构建最适模型。
4)输出各部分贡献度(R2),绘制载荷图。
4.1 模型构建
由于模型的拟合不足或过拟合都会对数据分析造成影响,因此在建立模型之前,我们会对模型进行预评估。模型的关联和正交部分包含一定数量的组分(component,类似PCA分析的主成分PC1、PC2)。我们采用交叉验证法(cross-validation)对每部分所包含的组分个数进行评估与设定。交叉验证法通过多次预建模,选择预测误差(prediction error)最小的模型进行后续分析。
| Model | nx | ny | n | prediction error |
|---|---|---|---|---|
| dif | 0 | 1 | 4 | 0.8159481 |
| LN-vs-LL | 0 | 0 | 4 | 0.8718620 |
| TN-vs-TL | 0 | 0 | 4 | 0.7448241 |
| LL-vs-TL | 0 | 0 | 3 | 0.9848046 |
| LN-vs-TN | 0 | 0 | 4 | 0.5221112 |
4.2 贡献度评估
贡献度是指模型各部分对总变异的解释程度,用R2表示。R2值越高,表示该部分对模型的解释能力越好。
| Model | R2X | R2Y | R2Xcorr | R2Ycorr |
|---|---|---|---|---|
| dif | 0.911 | 0.947 | 0.911 | 0.941 |
| LN-vs-LL | 0.978 | 0.988 | 0.978 | 0.988 |
| TN-vs-TL | 0.978 | 0.987 | 0.978 | 0.987 |
| LL-vs-TL | 0.934 | 0.981 | 0.934 | 0.981 |
| LN-vs-TN | 0.991 | 0.993 | 0.991 | 0.993 |
4.3 载荷图
为了查看哪些代谢物和基因相互关联,我们对joint部分的变量分别绘制不同组学的载荷图(loadings plot)。载荷值(loading value)表示变量(代谢物/基因)在各组分的解释能力(即对组间差异的贡献度),载荷值的正负表示与另一组学正关联或负相关;载荷值的绝对值越大,表示关联越强。
- dif
- LN-vs-LL
- TN-vs-TL
- LL-vs-TL
- LN-vs-TN
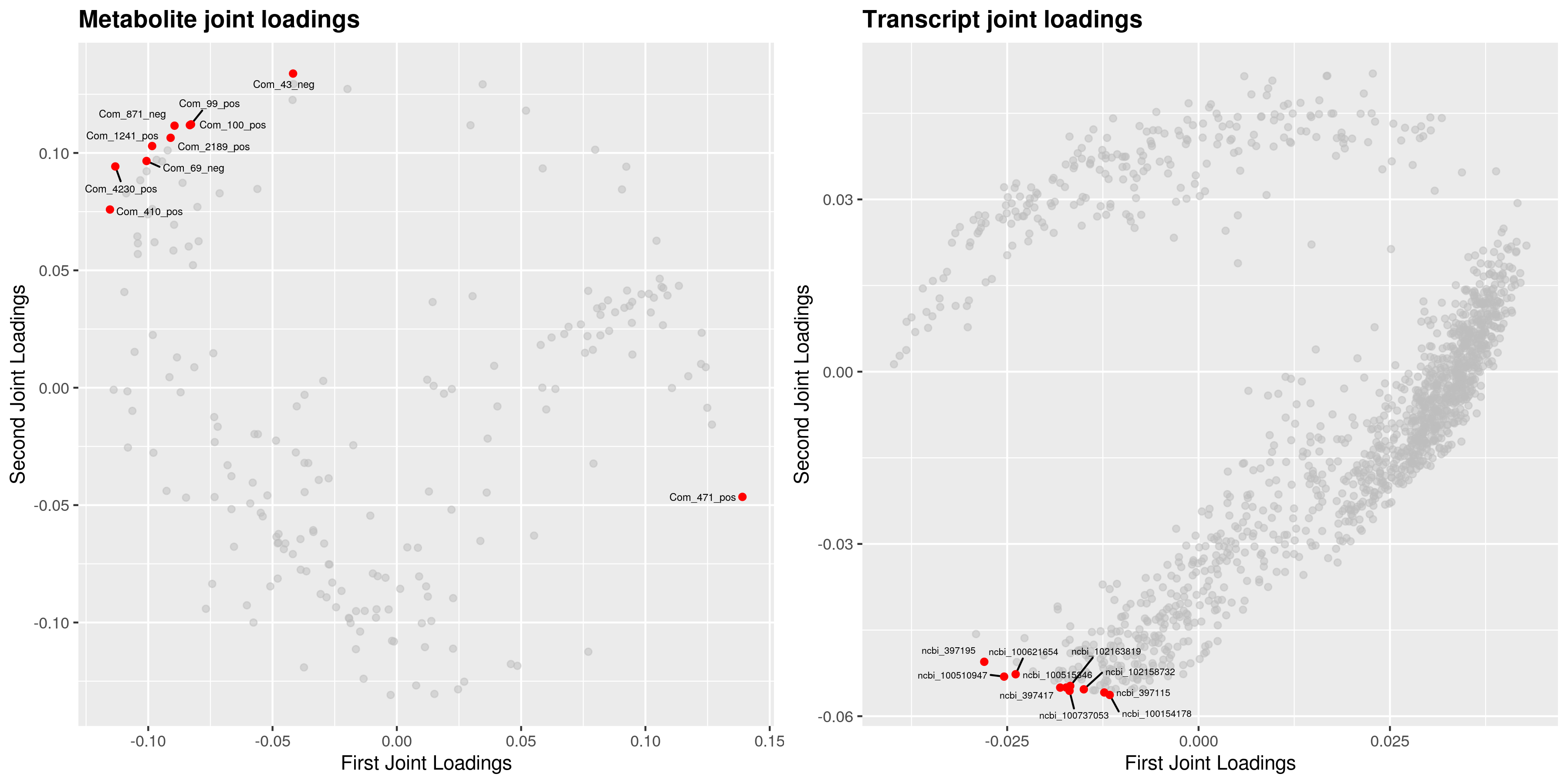
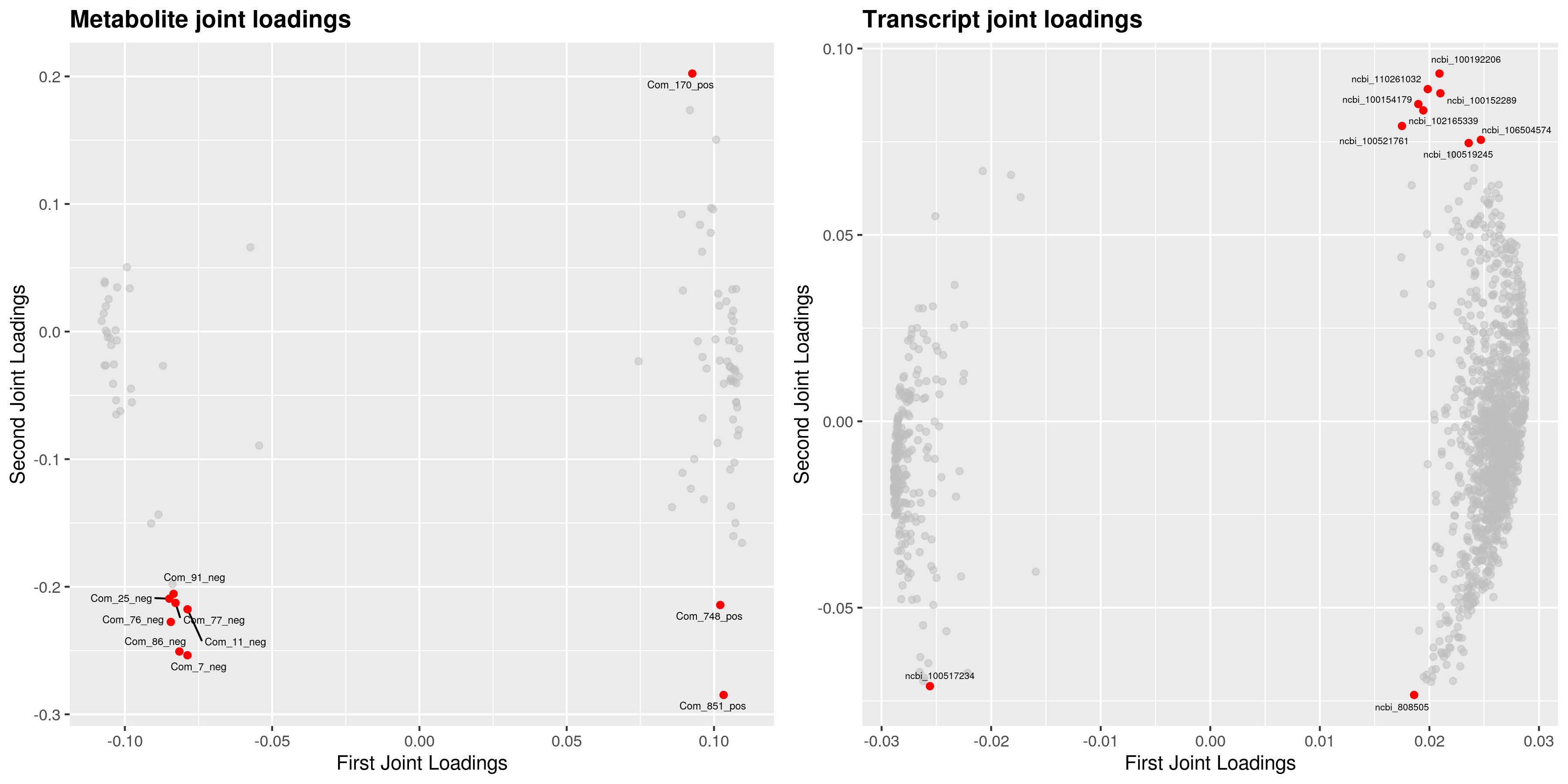
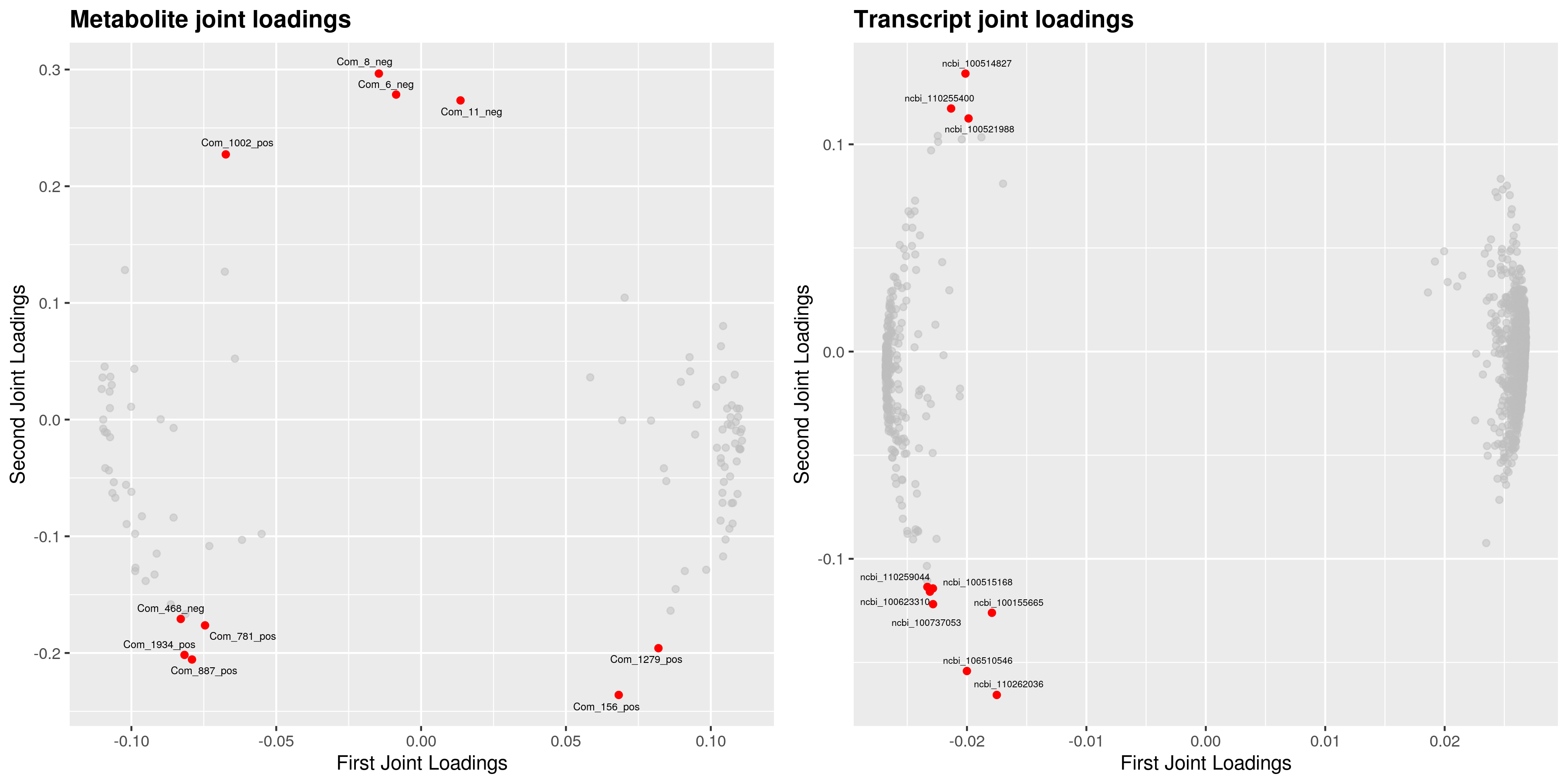
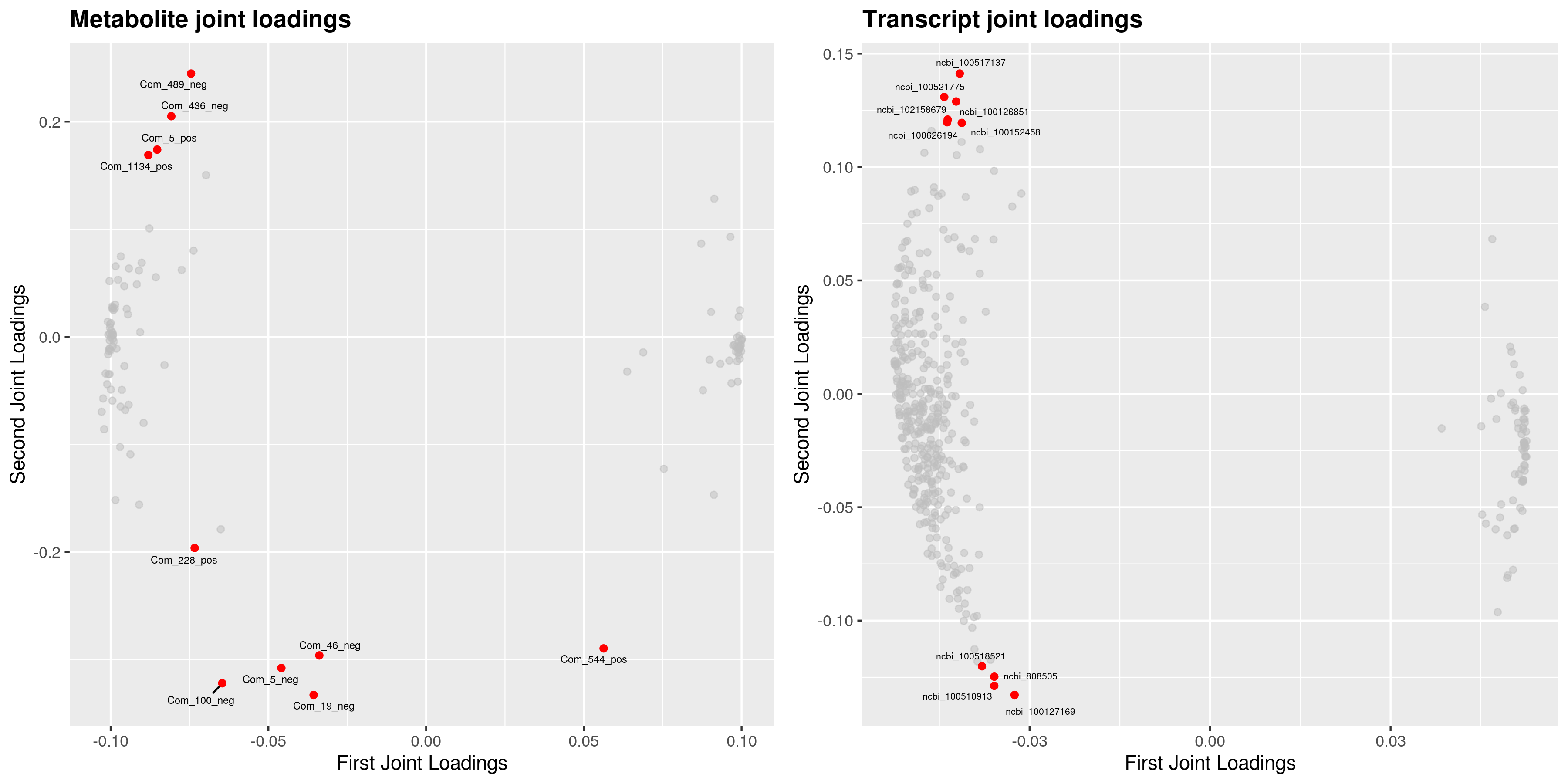
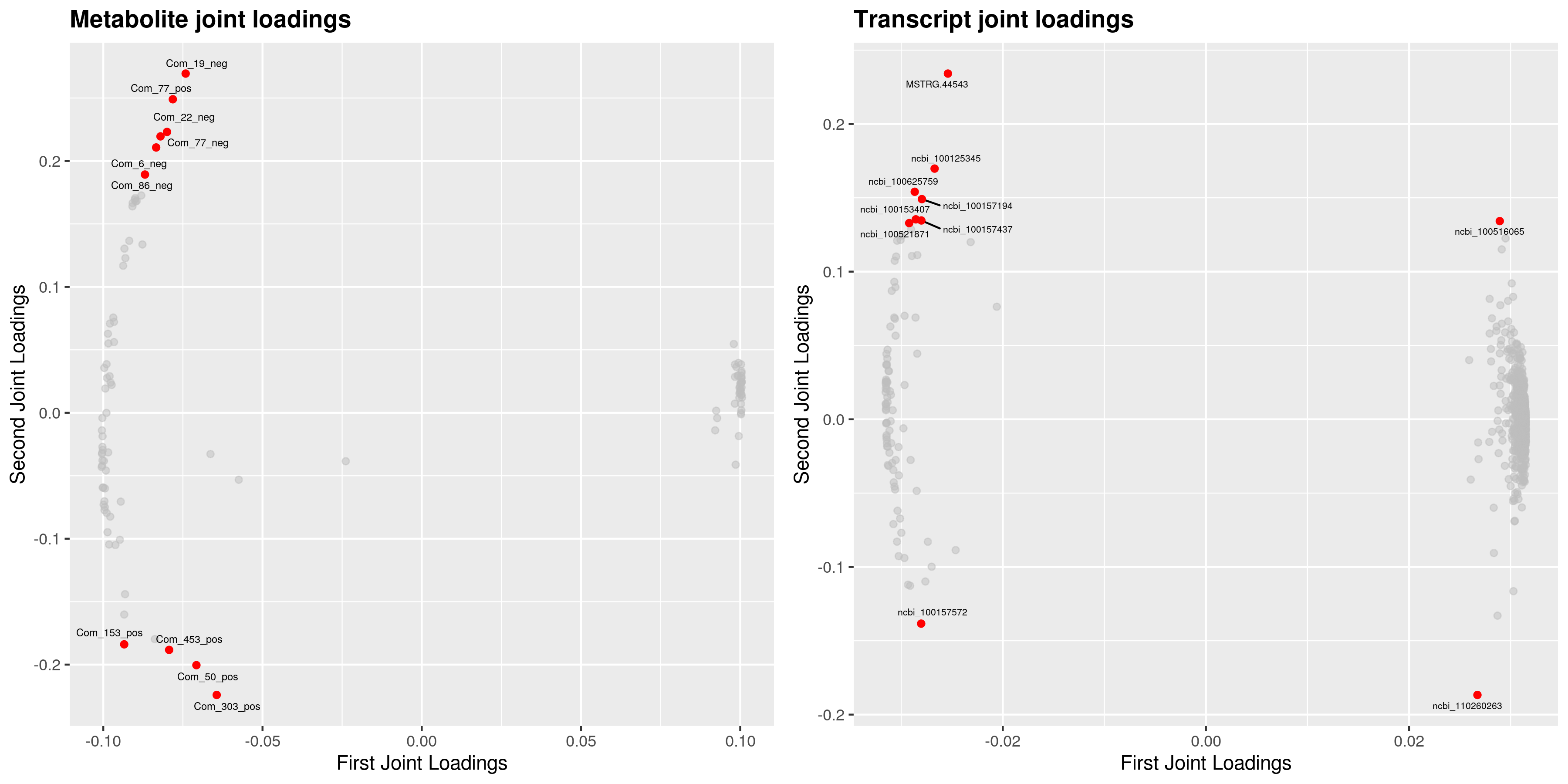
Fig 4-3-1 两组学O2PLS载荷图
4.4 组间元素关联分析
根据元素loading值结果,我们筛选出前两个维度loading值平方和前25的基因和代谢物进行整合绘制loading图,以展示关联程度最大的基因和代谢物。
差异代谢物与差异基因(dif)O2PLS分析loading值结果如下:
| Metabolite | Loading_1 | Loading_2 | MS2_name | MS1_name | formula | rtmin | mz | Class | CAS | HMDB | C_ID | Pathway | KEGG_B_class | KEGG_A_class |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Com_4230_pos | -0.113243860622071 | 0.0942462180558674 | 2-morpholinophenyl 3,5-dimethyl-4-isoxazolesulfonate | -- | C15 H18 N2 O5 S | 6.476 | 339.10037 | -- | -- | -- | - | - | - | - |
| Com_471_pos | 0.139021309893661 | -0.046495213512659 | Gly-Tyr | -- | C11 H14 N2 O4 | 3.322 | 239.10249 | Carboxylic acids and derivatives | 658-79-7 | HMDB0028853 | - | - | - | - |
| Com_871_neg | -0.0894203409471432 | 0.11161575990723 | N-(1,2,3,4-tetrahydro-1-naphthalenyl)benzenesulfonamide | -- | C16 H17 N O2 S | 2.684 | 286.09299 | -- | -- | -- | - | - | - | - |
| Com_1241_pos | -0.0984274411311528 | 0.102953405130596 | YMH | -- | C20 H27 N5 O5 S | 9.536 | 472.15707 | -- | -- | -- | - | - | - | - |
| Com_43_neg | -0.041743249129072 | 0.133817469512239 | Phenylacetylglycine | -- | C10 H11 N O3 | 7.014 | 192.06671 | Carboxylic acids and derivatives | 500-98-1 | HMDB0000821 | C05598 | ko00360//Phenylalanine metabolism | Amino acid metabolism | Metabolism |
| Com_2189_pos | -0.0909976052370658 | 0.106458156729543 | ��-Hydroxyalprazolam | -- | C17 H13 Cl N4 O | 8.957 | 325.08807 | -- | -- | -- | - | - | - | - |
| Com_69_neg | -0.10068276342123 | 0.09658402106037 | Oxoamide | -- | C10 H12 N2 O2 | 8.805 | 191.08255 | Organooxygen compounds | 713-05-3 | HMDB0001004 | - | - | - | - |
| Com_100_pos | -0.0832437303218206 | 0.111855671470694 | trans-3-Hydroxycotinine | -- | C10 H12 N2 O2 | 9.989 | 193.09712 | Pyridines and derivatives | 34834-67-8 | HMDB0001390 | - | - | - | - |
| Com_99_pos | -0.082789847058525 | 0.112164913550825 | Indole-3-acetic acid | -- | C10 H9 N O2 | 9.947 | 176.07037 | Indoles and derivatives | 87-51-4 | HMDB0000197 | C00954 | ko01100//Metabolic pathways;ko00380//Tryptophan metabolism | Amino acid metabolism;Global and overview maps | Metabolism |
| Com_410_pos | -0.115390834704968 | 0.0759082809782144 | 1-Phenyl-3-methyl-5-pyrazolone | -- | C10 H10 N2 O | 8.711 | 175.0865 | Azoles | 89-25-8 | HMDB0006240 | - | - | - | - |
| Com_254_pos | -0.0966368904264211 | 0.0970809007908701 | Pectolinarin | -- | C29 H34 O15 | 11.994 | 623.19794 | -- | -- | -- | - | - | - | - |
| Com_3450_pos | -0.092178194155333 | 0.101105525246011 | 2-(4-methoxyphenyl)-3-(2-phenyl-1H-imidazol-4-yl)acrylonitrile | -- | C19 H15 N3 O | 11.019 | 302.12579 | -- | -- | -- | - | - | - | - |
| Com_1315_pos | -0.108852838695884 | 0.0827923828388198 | WLK | -- | C23 H35 N5 O4 | 6.568 | 428.26154 | -- | -- | -- | - | - | - | - |
| Com_4489_pos | -0.100649749162475 | 0.0921987273517646 | 1-(4-methylphenyl)-3,5-diphenylpent-2-ene-1,5-dione | -- | C24 H20 O2 | 12.449 | 363.13657 | -- | -- | -- | - | - | - | - |
| Com_3927_pos | 0.0770228709739155 | -0.11246197351702 | Sulfamethazine | -- | C12 H14 N4 O2 S | 12.089 | 279.09302 | Benzene and substituted derivatives | 57-68-1 | HMDB0015522 | - | - | - | - |
| Com_1685_pos | -0.103209499567701 | 0.0883841028425779 | -- | 3-Methylindole | C9 H9 N | 8.674 | 132.08076 | Indoles and derivatives | 83-34-1 | HMDB0000466 | - | - | - | - |
| Com_2489_pos | -0.0415559426713497 | 0.12936676687919 | Cytidine | -- | C9 H13 N3 O5 | 1.518 | 244.09261 | Pyrimidine nucleosides | 65-46-3 | HMDB0000089 | C00475 | ko01100//Metabolic pathways;ko00240//Pyrimidine metabolism | Global and overview maps;Nucleotide metabolism | Metabolism |
| Com_2463_pos | -0.0944076320394717 | 0.0964203082149986 | 2-Isopropylmalic acid | -- | C7 H12 O5 | 1.356 | 194.10226 | Fatty Acyls | 3237-44-3 | HMDB0000402 | C02504 | ko01100//Metabolic pathways;ko01110//Biosynthesis of secondary metabolites;ko01230//Biosynthesis of amino acids;ko01210//2-Oxocarboxylic acid metabolism;ko00620//Pyruvate metabolism;ko00290//Valine, leucine and isoleucine biosynthesis | Amino acid metabolism;Carbohydrate metabolism;Global and overview maps | Metabolism |
| Com_1603_neg | 0.0345350429323179 | 0.129247748973283 | Guanosine-3',5'-cyclic monophosphate | -- | C10 H12 N5 O7 P | 2.056 | 344.03339 | -- | -- | -- | - | - | - | - |
| Com_511_pos | 0.0923090328262065 | 0.0941530442920312 | 6-(3-hydroxybutan-2-yl)-5-(hydroxymethyl)-4-methoxy-2H-pyran-2-one | -- | C11 H16 O5 | 2.308 | 267.05841 | -- | -- | -- | - | - | - | - |
| Com_429_pos | 0.0151211687906818 | -0.13044200192806 | DL-Arginine | -- | C6 H14 N4 O2 | 1.351 | 175.11885 | -- | -- | -- | - | - | - | - |
| Com_2878_pos | -0.00248748912616449 | -0.130862673867085 | HexCer-NS (d18:1/16:0) | -- | C40 H77 N O8 | 16.646 | 700.57056 | -- | -- | -- | - | - | - | - |
| Com_3609_pos | 0.0246237824160437 | -0.128473003107808 | Amoxicillin | -- | C16 H19 N3 O5 S | 6.032 | 388.09918 | Lactams | 26787-78-0 | HMDB0015193 | - | - | - | - |
| Com_303_pos | -0.0419441614955391 | 0.122586267497989 | L-Glutathione oxidized | -- | C20 H32 N6 O12 S2 | 2.218 | 613.1582 | -- | -- | -- | - | - | - | - |
| Com_53_neg | 0.0797892566818495 | 0.10136901610249 | Uridine | -- | C9 H12 N2 O6 | 2.369 | 243.06209 | Pyrimidine nucleosides | 58-96-8 | HMDB0000296 | C00299 | ko01100//Metabolic pathways;ko00240//Pyrimidine metabolism | Global and overview maps;Nucleotide metabolism | Metabolism |
| Transcript | Loading_1 | Loading_2 | Symbol | Description | KEGG_A_class | KEGG_B_class | Pathway | K_ID | GO.Component | GO.Function | GO.Process |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ncbi_100510947 | -0.0254054008430117 | -0.0531052391889094 | PTGDR2 | prostaglandin D2 receptor 2, transcript variant X1 | - | - | - | - | GO:0005887//integral component of plasma membrane;GO:0016020//membrane;GO:0016021//integral component of membrane;GO:0043005//neuron projection | GO:0001785//prostaglandin J receptor activity;GO:0004930//G protein-coupled receptor activity;GO:0004956//prostaglandin D receptor activity;GO:0004958//prostaglandin F receptor activity;GO:0042277//peptide binding;GO:0042923//neuropeptide binding | GO:0006935//chemotaxis;GO:0007165//signal transduction;GO:0007186//G protein-coupled receptor signaling pathway;GO:0007193//adenylate cyclase-inhibiting G protein-coupled receptor signaling pathway;GO:0007218//neuropeptide signaling pathway;GO:0019722//calcium-mediated signaling;GO:0045745//positive regulation of G protein-coupled receptor signaling pathway;GO:2000255//negative regulation of male germ cell proliferation |
| ncbi_100737053 | -0.016857417820817 | -0.0555806709745357 | PPP2R2C | protein phosphatase 2 regulatory subunit Bgamma, transcript variant X1 | Environmental Information Processing;Human Diseases;Cellular Processes;Environmental Information Processing;Organismal Systems;Organismal Systems;Human Diseases;Environmental Information Processing;Environmental Information Processing;Human Diseases;Genetic Information Processing | Signal transduction;Infectious diseases;Cellular community - eukaryotes;Signal transduction;Circulatory system;Nervous system;Infectious diseases;Signal transduction;Signal transduction;Infectious diseases;Translation | ko04151//PI3K-Akt signaling pathway;ko05165//Human papillomavirus infection;ko04530//Tight junction;ko04390//Hippo signaling pathway;ko04261//Adrenergic signaling in cardiomyocytes;ko04728//Dopaminergic synapse;ko05160//Hepatitis C;ko04152//AMPK signaling pathway;ko04071//Sphingolipid signaling pathway;ko05142//Chagas disease (American trypanosomiasis);ko03015//mRNA surveillance pathway | K04354;K04354;K04354;K04354;K04354;K04354;K04354;K04354;K04354;K04354;K04354 | GO:0000159//protein phosphatase type 2A complex;GO:0005829//cytosol | GO:0005515//protein binding;GO:0019888//protein phosphatase regulator activity | GO:0043666//regulation of phosphoprotein phosphatase activity;GO:0070262//peptidyl-serine dephosphorylation |
| ncbi_397417 | -0.0180761366834718 | -0.0550187975414674 | MT1A | metallothionein 1A | Organismal Systems | Digestive system | ko04978//Mineral absorption | K14739 | GO:0005623//cell;GO:0005634//nucleus;GO:0005737//cytoplasm;GO:0048471//perinuclear region of cytoplasm | GO:0008270//zinc ion binding;GO:0046872//metal ion binding | GO:0006882//cellular zinc ion homeostasis;GO:0010273//detoxification of copper ion;GO:0045926//negative regulation of growth;GO:0071276//cellular response to cadmium ion;GO:0071280//cellular response to copper ion;GO:0071294//cellular response to zinc ion |
| ncbi_100621654 | -0.0238966663605797 | -0.0526935463245403 | FAM3D | family with sequence similarity 3 member D, transcript variant X12 | - | - | - | - | GO:0016020//membrane;GO:0016021//integral component of membrane | - | GO:0070093//negative regulation of glucagon secretion |
| ncbi_397195 | -0.0279972181885951 | -0.0505241165969217 | ADM | adrenomedullin | - | - | - | - | GO:0005576//extracellular region;GO:0005615//extracellular space;GO:0005737//cytoplasm | GO:0005179//hormone activity;GO:0031700//adrenomedullin receptor binding | GO:0001570//vasculogenesis;GO:0001843//neural tube closure;GO:0002031//G protein-coupled receptor internalization;GO:0003073//regulation of systemic arterial blood pressure;GO:0007189//adenylate cyclase-activating G protein-coupled receptor signaling pathway;GO:0007507//heart development;GO:0008284//positive regulation of cell proliferation;GO:0010460//positive regulation of heart rate;GO:0031623//receptor internalization;GO:0035809//regulation of urine volume;GO:0043116//negative regulation of vascular permeability;GO:0045766//positive regulation of angiogenesis;GO:0045906//negative regulation of vasoconstriction;GO:0048589//developmental growth;GO:0060670//branching involved in labyrinthine layer morphogenesis;GO:0060712//spongiotrophoblast layer development;GO:0097084//vascular smooth muscle cell development;GO:0097647//amylin receptor signaling pathway;GO:1990410//adrenomedullin receptor signaling pathway;GO:2001214//positive regulation of vasculogenesis |
| ncbi_100515346 | -0.0172857975324992 | -0.0550023250887469 | ACKR3 | atypical chemokine receptor 3 | Environmental Information Processing | Signaling molecules and interaction | ko04060//Cytokine-cytokine receptor interaction | K04304 | GO:0005623//cell;GO:0005768//endosome;GO:0005886//plasma membrane;GO:0005905//clathrin-coated pit;GO:0009897//external side of plasma membrane;GO:0009986//cell surface;GO:0016020//membrane;GO:0016021//integral component of membrane;GO:0043231//intracellular membrane-bounded organelle | GO:0004930//G protein-coupled receptor activity;GO:0005044//scavenger receptor activity;GO:0015026//coreceptor activity;GO:0016493//C-C chemokine receptor activity;GO:0016494//C-X-C chemokine receptor activity;GO:0019956//chemokine binding;GO:0019957//C-C chemokine binding;GO:0019958//C-X-C chemokine binding | GO:0001525//angiogenesis;GO:0001570//vasculogenesis;GO:0006935//chemotaxis;GO:0006955//immune response;GO:0007165//signal transduction;GO:0007186//G protein-coupled receptor signaling pathway;GO:0007204//positive regulation of cytosolic calcium ion concentration;GO:0019722//calcium-mediated signaling;GO:0031623//receptor internalization;GO:0060326//cell chemotaxis;GO:0070098//chemokine-mediated signaling pathway;GO:0070374//positive regulation of ERK1 and ERK2 cascade;GO:1902230//negative regulation of intrinsic apoptotic signaling pathway in response to DNA damage |
| ncbi_100154178 | -0.011605704099471 | -0.0562984310812665 | NEDD9 | neural precursor cell expressed, developmentally down-regulated 9, transcript variant X1 | - | - | - | - | GO:0000922//spindle pole;GO:0005634//nucleus;GO:0005654//nucleoplasm;GO:0005737//cytoplasm;GO:0005829//cytosol;GO:0005886//plasma membrane | GO:0005515//protein binding;GO:1990782//protein tyrosine kinase binding | GO:0007169//transmembrane receptor protein tyrosine kinase signaling pathway;GO:0016477//cell migration;GO:0030335//positive regulation of cell migration;GO:0061098//positive regulation of protein tyrosine kinase activity;GO:0090527//actin filament reorganization;GO:0090630//activation of GTPase activity;GO:1900026//positive regulation of substrate adhesion-dependent cell spreading |
| ncbi_102158732 | -0.01499091685618 | -0.0553190782599183 | FXYD6 | FXYD domain containing ion transport regulator 6, transcript variant X1 | - | - | - | - | GO:0016020//membrane;GO:0016021//integral component of membrane | GO:0017080//sodium channel regulator activity;GO:0099106//ion channel regulator activity | GO:0006811//ion transport;GO:0043269//regulation of ion transport;GO:2000649//regulation of sodium ion transmembrane transporter activity |
| ncbi_102163819 | -0.0167653255151326 | -0.054730658028349 | MS4A10 | membrane spanning 4-domains A10, transcript variant X1 | - | - | - | - | GO:0016020//membrane;GO:0016021//integral component of membrane | - | - |
| ncbi_397115 | -0.0123182989734467 | -0.05586759958117 | ARG1 | arginase 1 | Metabolism;Human Diseases;Metabolism;Metabolism;Metabolism | Global and overview maps;Infectious diseases;Global and overview maps;Amino acid metabolism;Amino acid metabolism | ko01100//Metabolic pathways;ko05146//Amoebiasis;ko01230//Biosynthesis of amino acids;ko00330//Arginine and proline metabolism;ko00220//Arginine biosynthesis | K01476;K01476;K01476;K01476;K01476 | GO:0005615//extracellular space;GO:0005737//cytoplasm;GO:0005829//cytosol | GO:0004053//arginase activity;GO:0016787//hydrolase activity;GO:0016813//hydrolase activity, acting on carbon-nitrogen (but not peptide) bonds, in linear amidines;GO:0030145//manganese ion binding;GO:0046872//metal ion binding | GO:0000050//urea cycle;GO:0006525//arginine metabolic process;GO:0019547//arginine catabolic process to ornithine;GO:0042130//negative regulation of T cell proliferation;GO:0042832//defense response to protozoan;GO:0046007//negative regulation of activated T cell proliferation;GO:0060336//negative regulation of interferon-gamma-mediated signaling pathway;GO:0070965//positive regulation of neutrophil mediated killing of fungus;GO:2000552//negative regulation of T-helper 2 cell cytokine production |
| ncbi_100519355 | -0.0137648035256135 | -0.0554794719196159 | F2RL2 | coagulation factor II thrombin receptor like 2 | Environmental Information Processing;Organismal Systems | Signaling molecules and interaction;Immune system | ko04080//Neuroactive ligand-receptor interaction;ko04610//Complement and coagulation cascades | K04235;K04235 | GO:0005623//cell;GO:0005887//integral component of plasma membrane;GO:0016020//membrane;GO:0016021//integral component of membrane;GO:0016324//apical plasma membrane;GO:0032991//protein-containing complex | GO:0004930//G protein-coupled receptor activity;GO:0015057//thrombin-activated receptor activity | GO:0007165//signal transduction;GO:0007186//G protein-coupled receptor signaling pathway;GO:0007596//blood coagulation;GO:0035025//positive regulation of Rho protein signal transduction;GO:0051482//positive regulation of cytosolic calcium ion concentration involved in phospholipase C-activating G protein-coupled signaling pathway;GO:0070493//thrombin-activated receptor signaling pathway |
| ncbi_100620701 | -0.0135341005622964 | -0.0554949909155963 | ECM1 | extracellular matrix protein 1, transcript variant X1 | - | - | - | - | GO:0005576//extracellular region;GO:0005615//extracellular space;GO:0062023//collagen-containing extracellular matrix | GO:0002020//protease binding;GO:0005134//interleukin-2 receptor binding;GO:0008022//protein C-terminus binding;GO:0019899//enzyme binding;GO:0043236//laminin binding | GO:0001938//positive regulation of endothelial cell proliferation;GO:0001960//negative regulation of cytokine-mediated signaling pathway;GO:0002063//chondrocyte development;GO:0002828//regulation of type 2 immune response;GO:0003416//endochondral bone growth;GO:0006357//regulation of transcription by RNA polymerase II;GO:0006954//inflammatory response;GO:0007165//signal transduction;GO:0010466//negative regulation of peptidase activity;GO:0030500//regulation of bone mineralization;GO:0030502//negative regulation of bone mineralization;GO:0045766//positive regulation of angiogenesis;GO:2000404//regulation of T cell migration |
| ncbi_100157572 | -0.0188364773680572 | -0.053791734918358 | GALNT15 | polypeptide N-acetylgalactosaminyltransferase 15 | Metabolism;Metabolism | Global and overview maps;Glycan biosynthesis and metabolism | ko01100//Metabolic pathways;ko00512//Mucin type O-glycan biosynthesis | K00710;K00710 | GO:0000139//Golgi membrane;GO:0005794//Golgi apparatus;GO:0016020//membrane;GO:0016021//integral component of membrane;GO:0030133//transport vesicle | GO:0016740//transferase activity;GO:0016757//transferase activity, transferring glycosyl groups;GO:0030246//carbohydrate binding | GO:0006486//protein glycosylation |
| ncbi_100144490 | 0.0227614442359746 | 0.0519151399685711 | IGFBP4 | insulin like growth factor binding protein 4 | - | - | - | - | GO:0005576//extracellular region;GO:0005615//extracellular space | GO:0005520//insulin-like growth factor binding;GO:0031994//insulin-like growth factor I binding;GO:0031995//insulin-like growth factor II binding | GO:0001558//regulation of cell growth;GO:0010906//regulation of glucose metabolic process;GO:0040008//regulation of growth;GO:0043410//positive regulation of MAPK cascade;GO:0043567//regulation of insulin-like growth factor receptor signaling pathway;GO:0043568//positive regulation of insulin-like growth factor receptor signaling pathway;GO:0044342//type B pancreatic cell proliferation |
| ncbi_106509958 | -0.0114308285536688 | -0.0554470744577443 | MTSS1 | MTSS1, I-BAR domain containing, transcript variant X1 | - | - | - | - | - | GO:0003779//actin binding | GO:0007009//plasma membrane organization;GO:0030036//actin cytoskeleton organization |
| ncbi_397566 | -0.0183189818211 | -0.0534487530012847 | PGM1 | phosphoglucomutase 1 | Metabolism;Metabolism;Metabolism;Metabolism;Metabolism;Metabolism;Metabolism | Global and overview maps;Nucleotide metabolism;Carbohydrate metabolism;Carbohydrate metabolism;Carbohydrate metabolism;Carbohydrate metabolism;Carbohydrate metabolism | ko01100//Metabolic pathways;ko00230//Purine metabolism;ko00010//Glycolysis / Gluconeogenesis;ko00520//Amino sugar and nucleotide sugar metabolism;ko00500//Starch and sucrose metabolism;ko00052//Galactose metabolism;ko00030//Pentose phosphate pathway | K01835;K01835;K01835;K01835;K01835;K01835;K01835 | GO:0005829//cytosol | GO:0000287//magnesium ion binding;GO:0004614//phosphoglucomutase activity;GO:0016868//intramolecular transferase activity, phosphotransferases;GO:0046872//metal ion binding | GO:0005975//carbohydrate metabolic process;GO:0005978//glycogen biosynthetic process;GO:0006006//glucose metabolic process;GO:0019388//galactose catabolic process;GO:0071704//organic substance metabolic process |
| ncbi_106510284 | -0.0216268779395072 | -0.0521847908039349 | LOC106510284 | apolipoprotein L3-like, transcript variant X1 | - | - | - | - | GO:0005576//extracellular region;GO:0005615//extracellular space;GO:0016020//membrane;GO:0016021//integral component of membrane | GO:0008289//lipid binding | GO:0006869//lipid transport;GO:0042157//lipoprotein metabolic process |
| ncbi_100154641 | -0.010827812970905 | -0.0552417969605562 | ABI3BP | ABI family member 3 binding protein, transcript variant X1 | - | - | - | - | - | GO:0005515//protein binding | - |
| ncbi_100157437 | -0.0121755678428262 | -0.0548716626985484 | RGS4 | regulator of G protein signaling 4 | - | - | - | - | GO:0005634//nucleus;GO:0005737//cytoplasm | GO:0005096//GTPase activator activity | GO:0007186//G protein-coupled receptor signaling pathway;GO:0043547//positive regulation of GTPase activity |
| ncbi_100525009 | -0.0170287176448306 | -0.0535474025446255 | ZNF395 | zinc finger protein 395, transcript variant 1 | - | - | - | - | GO:0005634//nucleus;GO:0005654//nucleoplasm;GO:0005737//cytoplasm;GO:0005829//cytosol | GO:0000978//RNA polymerase II proximal promoter sequence-specific DNA binding;GO:0000987//proximal promoter sequence-specific DNA binding;GO:0003676//nucleic acid binding;GO:0003677//DNA binding;GO:0005515//protein binding | GO:0006357//regulation of transcription by RNA polymerase II |
| ncbi_102162371 | -0.0128564723912642 | -0.0546035831341483 | FBLN7 | fibulin 7 | - | - | - | - | - | GO:0005509//calcium ion binding | - |
| ncbi_396775 | -0.00875354792375368 | -0.0553816386529524 | TIMP3 | TIMP metallopeptidase inhibitor 3 | Human Diseases;Human Diseases | Cancers;Cancers | ko05205//Proteoglycans in cancer;ko05206//MicroRNAs in cancer | K16866;K16866 | GO:0005576//extracellular region;GO:0005604//basement membrane;GO:0005615//extracellular space;GO:0031012//extracellular matrix | GO:0002020//protease binding;GO:0005515//protein binding;GO:0008191//metalloendopeptidase inhibitor activity | GO:0009725//response to hormone;GO:0010033//response to organic substance;GO:0010951//negative regulation of endopeptidase activity;GO:0034097//response to cytokine;GO:0051045//negative regulation of membrane protein ectodomain proteolysis;GO:0070373//negative regulation of ERK1 and ERK2 cascade;GO:0071310//cellular response to organic substance;GO:1903984//positive regulation of TRAIL-activated apoptotic signaling pathway;GO:1904684//negative regulation of metalloendopeptidase activity |
| ncbi_396877 | -0.0237446042805543 | -0.0505703311769263 | CFD | complement factor D | Organismal Systems;Human Diseases | Immune system;Infectious diseases | ko04610//Complement and coagulation cascades;ko05150//Staphylococcus aureus infection | K01334;K01334 | GO:0005576//extracellular region;GO:0005615//extracellular space | GO:0004252//serine-type endopeptidase activity;GO:0008233//peptidase activity;GO:0008236//serine-type peptidase activity;GO:0016787//hydrolase activity | GO:0002376//immune system process;GO:0006508//proteolysis;GO:0006957//complement activation, alternative pathway;GO:0007219//Notch signaling pathway;GO:0009617//response to bacterium;GO:0045087//innate immune response |
| ncbi_100153407 | -0.0177987465719213 | -0.0529327073648918 | CSDC2 | cold shock domain containing C2 | - | - | - | - | GO:0005737//cytoplasm | GO:0003676//nucleic acid binding;GO:0003730//mRNA 3'-UTR binding | GO:0043488//regulation of mRNA stability |
| ncbi_396670 | -0.00840727947082502 | -0.0551246758201098 | SCD | stearoyl-CoA desaturase | Environmental Information Processing;Organismal Systems;Metabolism;Metabolism | Signal transduction;Endocrine system;Global and overview maps;Lipid metabolism | ko04152//AMPK signaling pathway;ko03320//PPAR signaling pathway;ko01212//Fatty acid metabolism;ko01040//Biosynthesis of unsaturated fatty acids | K00507;K00507;K00507;K00507 | GO:0016020//membrane;GO:0016021//integral component of membrane | GO:0016491//oxidoreductase activity;GO:0016717//oxidoreductase activity, acting on paired donors, with oxidation of a pair of donors resulting in the reduction of molecular oxygen to two molecules of water | GO:0006629//lipid metabolic process;GO:0006633//fatty acid biosynthetic process;GO:0055114//oxidation-reduction process |
- dif
- LN-vs-LL
- TN-vs-TL
- LL-vs-TL
- LN-vs-TN
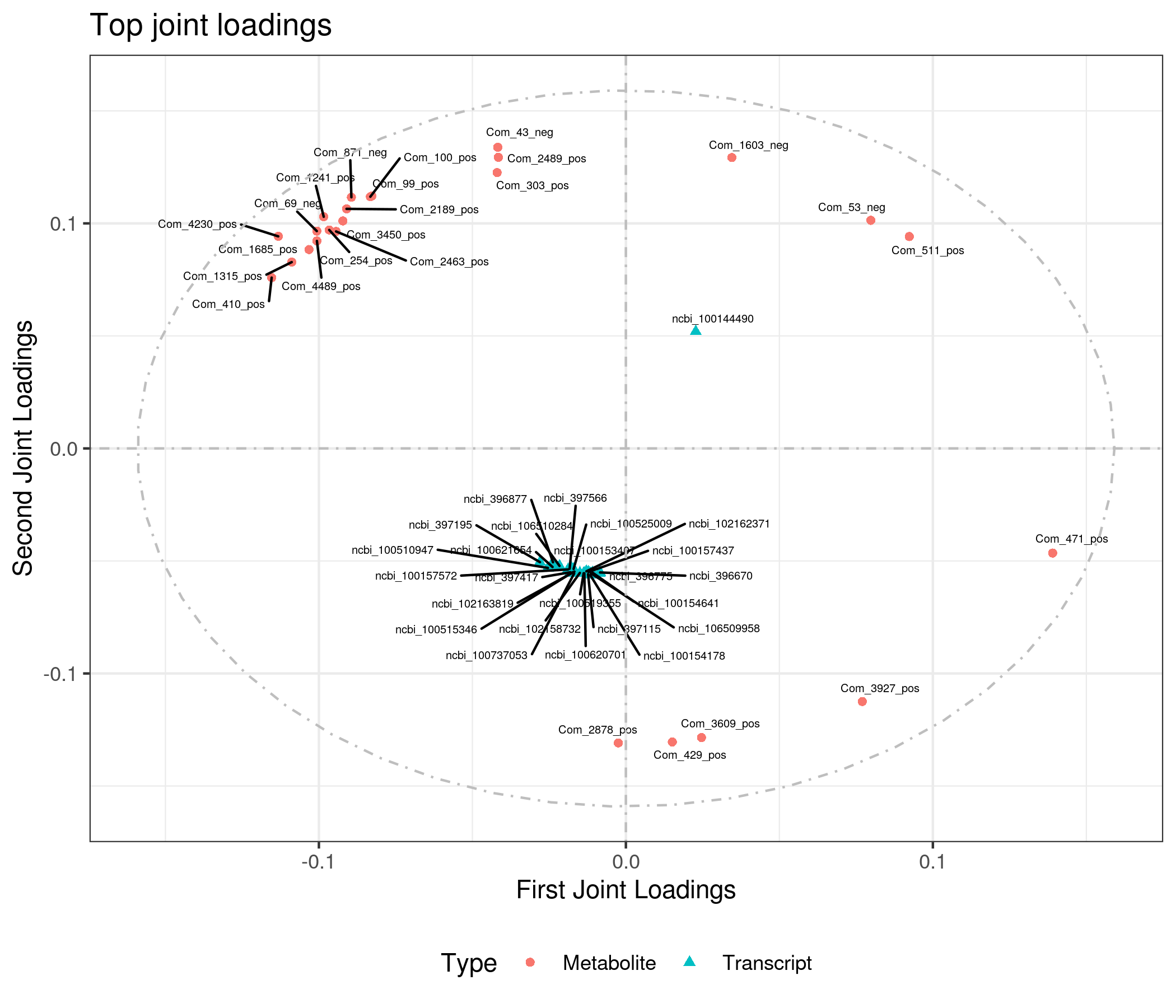
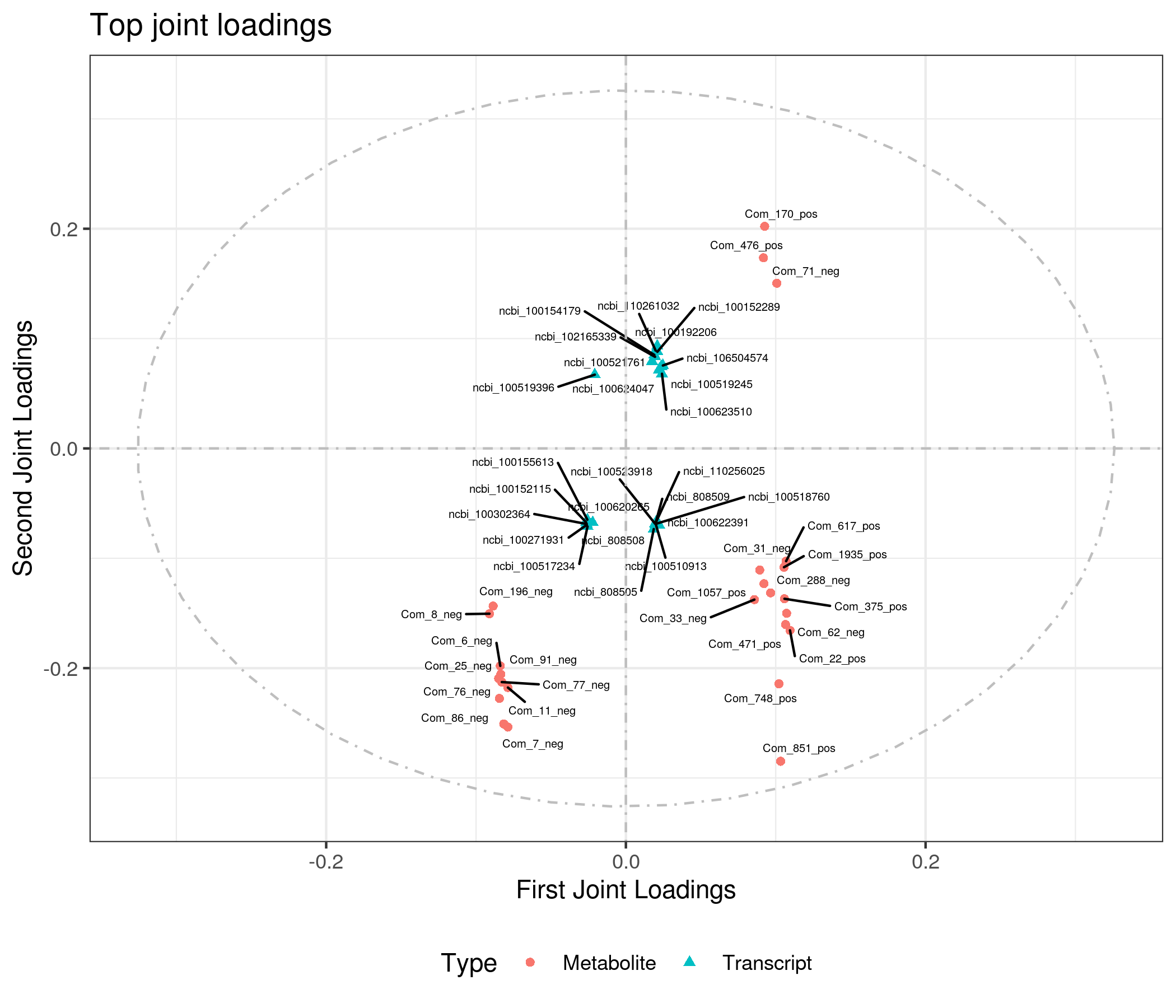

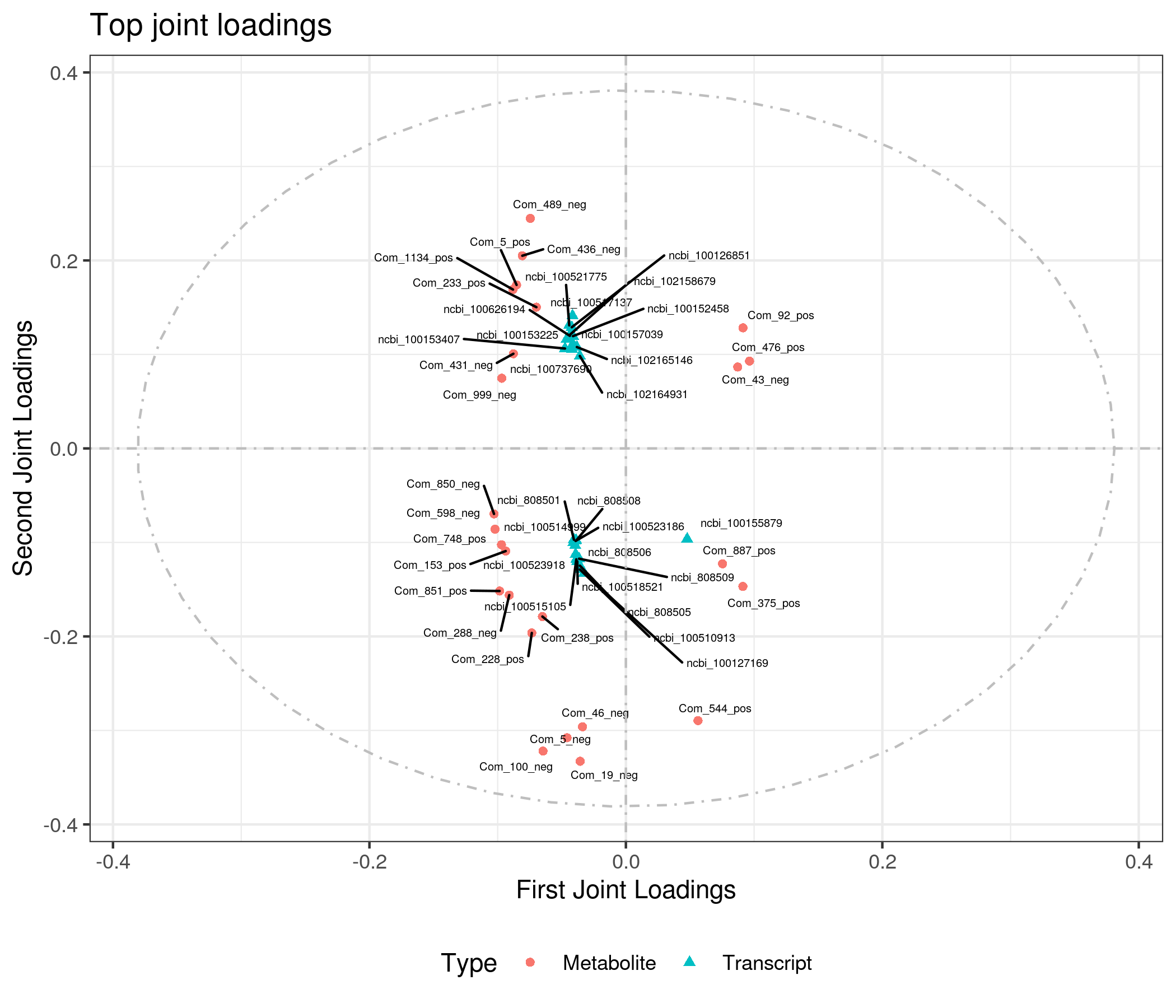
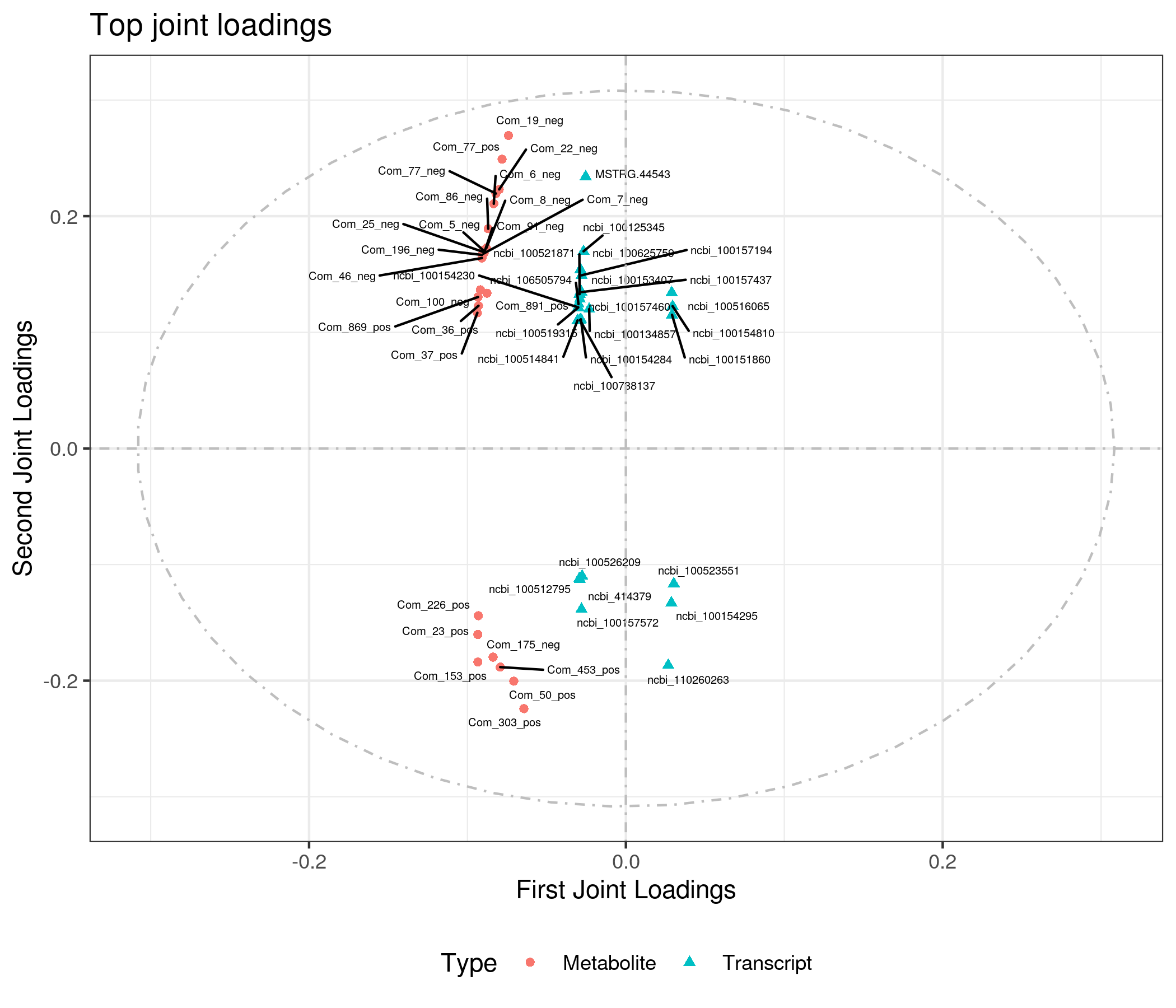
Fig 4-4-1 两组学关联载荷图
5 相关性系数模型
5.1 Pearson系数计算
皮尔逊相关系数(Pearson correlation coefficient)可用来度量两个变量之间的相互关系,代表了两个变量共变性的强弱,取值范围为[-1,+1]。计算基因表达量与代谢物丰度的pearson系数,以评估基因与代谢物的相关性[6][7][8]。
包含两种类型:
1)所有差异基因(各比较组间差异基因的并集)表达量与所有差异代谢物(各比较组间差异代谢物的并集)丰度的pearson系数;
2)所有差异基因(各比较组间差异基因的并集)表达量与所有代谢物丰度的pearson系数。
具体分析结果如下:
| 关联类型 | 基因数据来源 | 代谢物数据来源 | 系数列表 | 热图 |
|---|---|---|---|---|
| 差异基因与 差异代谢物 | 转录组差异集合 | 代谢组差异集合 | dif.pearson.xls | dif.heatmap.corMatrix.png |
| 差异基因与 所有代谢物 | 转录组差异集合 | 所有代谢物 | dif_all.pearson.xls | dif_all.top250.heatmap.png |
差异基因并集与差异代谢物peason相关性结果如下:
| Metabolite,Transcript | Gene_pathwayID | Metabolite | Metabolite_pathwayID | cor | p_value |
|---|---|---|---|---|---|
| ncbi_397323 | K19719;04151,04510,04512,04974,05165 | Com_99_pos | C00954;00380,01100 | 0.99573790797749 | 1.0997074369041e-11 |
| ncbi_397323 | K19719;04151,04510,04512,04974,05165 | Com_100_pos | -- | 0.995122243431639 | 2.15683664052079e-11 |
| ncbi_397323 | K19719;04151,04510,04512,04974,05165 | Com_871_neg | -- | 0.994015934997718 | 5.98267926095194e-11 |
| ncbi_100623369 | K06529;04360 | Com_938_pos | -- | 0.993440768667755 | 9.4571794963713e-11 |
| ncbi_110256379 | - | Com_3450_pos | -- | 0.992577791327635 | 1.75200919851018e-10 |
| ncbi_100286873 | K00355;00130,05200,05225,05418 | Com_3450_pos | -- | 0.991960249327033 | 2.60996794177091e-10 |
| ncbi_396769 | K01394;04657,04668,05202,05215,05323 | Com_62_neg | C00252;00500 | 0.991874059907596 | 2.75250281539745e-10 |
| ncbi_100523665 | - | Com_43_neg | C05598;00360 | 0.991828050092155 | 2.83109708976583e-10 |
| ncbi_110256379 | - | Com_254_pos | -- | 0.99171732160051 | 3.02760963458087e-10 |
| ncbi_100739325 | K07360;04014,04064,04650,04658,04659,04660,05340 | Com_5636_pos | -- | 0.991699661646902 | 3.05993387972836e-10 |
| ncbi_100515363 | K12326;04010,04014 | Com_2935_pos | -- | 0.991659309978524 | 3.13482763249513e-10 |
| ncbi_396769 | K01394;04657,04668,05202,05215,05323 | Com_617_pos | C00492;00052,02010 | 0.99164615703245 | 3.15955373922498e-10 |
| ncbi_110261555 | K06521;04360 | Com_6471_pos | -- | 0.991496885517343 | 3.45124624550085e-10 |
| ncbi_100286873 | K00355;00130,05200,05225,05418 | Com_2189_pos | -- | 0.991449805080897 | 3.54757596706865e-10 |
| ncbi_110256218 | - | Com_2935_pos | -- | 0.991437347936759 | 3.57342002099186e-10 |
| ncbi_100625219 | K05901;00740,00860,01100 | Com_2189_pos | -- | 0.991079452541301 | 4.38268444488785e-10 |
| ncbi_100623369 | K06529;04360 | Com_4321_pos | C02470;00380 | 0.99088875704156 | 4.87003737814589e-10 |
| ncbi_100621901 | K16904;00240,01100 | Com_43_neg | C05598;00360 | 0.990498038672161 | 6.00380574537078e-10 |
| ncbi_100737690 | - | Com_99_pos | C00954;00380,01100 | 0.99048355957112 | 6.04954188077389e-10 |
| ncbi_100155948 | - | Com_1036_neg | C02305;00330,01100 | 0.990450365825317 | 6.15544396594495e-10 |
| ncbi_100739325 | K07360;04014,04064,04650,04658,04659,04660,05340 | Com_6471_pos | -- | 0.990091416225014 | 7.39814523013613e-10 |
| ncbi_100626904 | K04386;04010,04060,04064,04380,04640,04659,04750,05146,05163,05166,05418 | Com_511_pos | -- | 0.989881835933655 | 8.21147605510647e-10 |
| ncbi_100286873 | K00355;00130,05200,05225,05418 | Com_1241_pos | -- | 0.989226452233044 | 1.12261302184965e-09 |
| ncbi_397323 | K19719;04151,04510,04512,04974,05165 | Com_2189_pos | -- | 0.989216589769261 | 1.12774221083208e-09 |
| ncbi_100737690 | - | Com_100_pos | -- | 0.989043139427971 | 1.22105100799898e-09 |
| ncbi_110261555 | K06521;04360 | Com_986_neg | -- | 0.988965815801127 | 1.26458514025912e-09 |
| ncbi_692156 | K01689;00010,01100,01200,01230,03018,04066 | Com_99_pos | C00954;00380,01100 | 0.98882204144304 | 1.34882278194787e-09 |
| ncbi_100511282 | K14958;04014,04625,05152 | Com_43_neg | C05598;00360 | 0.988739756924214 | 1.39901197198018e-09 |
| ncbi_100511285 | - | Com_3450_pos | -- | 0.988462164012995 | 1.57943807500204e-09 |
| ncbi_100625457 | - | Com_43_neg | C05598;00360 | 0.988277018875844 | 1.70976537500809e-09 |
| ncbi_396769 | K01394;04657,04668,05202,05215,05323 | Com_140_pos | C00208;00500,01100,02010,04742,04973 | 0.988026209434229 | 1.89986224688306e-09 |
| ncbi_396769 | K01394;04657,04668,05202,05215,05323 | Com_2085_pos | C00931;00860,01100,01110,01120 | 0.987896945435662 | 2.0042172034861e-09 |
| ncbi_100625219 | K05901;00740,00860,01100 | Com_1241_pos | -- | 0.987700329168024 | 2.1716723105646e-09 |
| ncbi_100154230 | K07997;04928 | Com_201_pos | -- | 0.987242885786697 | 2.60469198123594e-09 |
| ncbi_100152603 | K00699;00040,00053,00140,00830,00860,00980,00982,00983,01100,05204 | Com_43_neg | C05598;00360 | 0.987126921214098 | 2.72472038535937e-09 |
| ncbi_397614 | K05071;04060,04151,04630,04640,04658,04659,05200,05321 | Com_43_neg | C05598;00360 | 0.98709556511792 | 2.75792180685396e-09 |
| ncbi_692156 | K01689;00010,01100,01200,01230,03018,04066 | Com_100_pos | -- | 0.986941187339627 | 2.92612690427839e-09 |
| ncbi_110261555 | K06521;04360 | Com_5636_pos | -- | 0.986830924223538 | 3.05120207458661e-09 |
| ncbi_110256870 | - | Com_1603_neg | -- | -0.986555808997098 | 3.38195409399734e-09 |
| ncbi_100156071 | - | Com_1603_neg | -- | -0.986538564680428 | 3.40360095397079e-09 |
| ncbi_106510480 | K04746;00533 | Com_99_pos | C00954;00380,01100 | 0.986487649424929 | 3.46816116349112e-09 |
| ncbi_396769 | K01394;04657,04668,05202,05215,05323 | Com_98_neg | C01835;02010,04973 | 0.986302822685378 | 3.71078386574945e-09 |
| ncbi_396769 | K01394;04657,04668,05202,05215,05323 | Com_2662_pos | -- | 0.986288811624173 | 3.72971428041864e-09 |
| ncbi_396769 | K01394;04657,04668,05202,05215,05323 | Com_1105_pos | C00762;00140,01100,04960,05200,05215 | 0.986255280859366 | 3.77533093879632e-09 |
| ncbi_100155736 | K22284;00561,01100,04979 | Com_1603_neg | -- | 0.986043469948097 | 4.07388557282788e-09 |
| ncbi_106510480 | K04746;00533 | Com_100_pos | -- | 0.985919578182247 | 4.25705951245116e-09 |
| ncbi_654406 | - | Com_99_pos | C00954;00380,01100 | 0.985891029091357 | 4.30018666469728e-09 |
| ncbi_100037293 | K06751;04144,04145,04218,04514,04612,04650,04940,05163,05165,05166,05167,05168,05169,05203,05320,05330,05332,05416 | Com_6471_pos | -- | 0.985724441279942 | 4.55884636618179e-09 |
| ncbi_100037293 | K06751;04144,04145,04218,04514,04612,04650,04940,05163,05165,05166,05167,05168,05169,05203,05320,05330,05332,05416 | Com_986_neg | -- | 0.985562866319963 | 4.82143901010568e-09 |
| ncbi_100514576 | - | Com_1603_neg | -- | -0.985561252146159 | 4.82412191802372e-09 |
5.2 相关性热图
由于差异基因的数目普遍较多,不利于可视化展示关联特征,故先对所有差异基因进行筛选。取相关系数排名前50的差异基因与差异代谢物的相关性以热图展示:
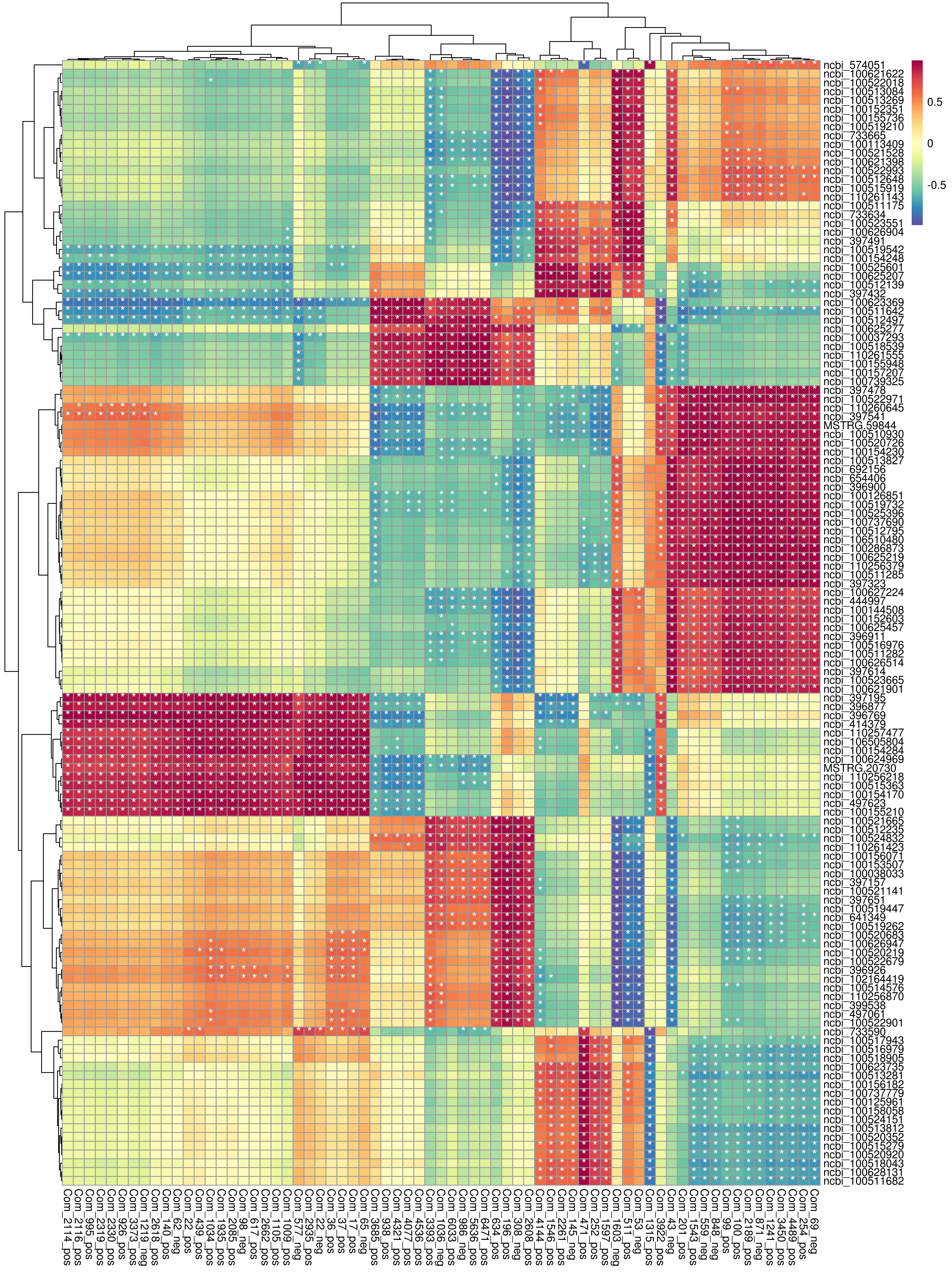 |
| Fig 5-2-1 基因表达量与代谢物丰度相关性热图图 |
5.3 相关性网络图
网络图可以展现处于重要关联位置的基因或代谢物,筛选出相关系数绝对值大于0.5且排名前250的差异基因与差异代谢物数据绘制网络图结果: pearson.sigma_net.html
 |
| Fig 5-3-1 基因表达量与代谢物丰度相关性网络图 |
6 目录结构
├── 01.expression_data [组学丰度数据]
│ ├── groups.txt [分组文件]
│ ├── met.diff.xls [代谢组差异并集丰度]
│ └── rna.diff.xls [转录组差异并集丰度]
├── 02.pathway [pathway模型结果目录]
│ ├── 1.dif_pathway [差异基因与差异代谢物的分析]
│ │ └── Z-vs-ZJ [比较组Z与ZJ的分析]
│ │ ├── Z-vs-ZJ.htm [pathway富集分析]
│ │ ├── Z-vs-ZJ_map [pathway图文件]
│ │ └── Z-vs-ZJ.path.xls [pathway注释]
│ ├── 2.dif_all_pathway [差异基因与所有代谢物的分析]
│ │ └── ZR-vs-ZJR_all [ZR与ZJR差异基因并集与所有代谢物分析]
│ │ ├── ZR-vs-ZJR_all.htm [pathway富集分析]
│ │ ├── ZR-vs-ZJR_all_map [pathway图文件]
│ │ └── ZR-vs-ZJR_all.path.xls [pathway注释]
│ └── 3.all_pathway [所有基因与所有代谢物的分析]
│ ├── all.htm [pathway富集分析]
│ ├── all_map [pathway图文件]
│ └── all.path.xls [pathway注释]
├── 03.o2pls [O2PLS模型结果目录]
│ ├── all.cv.stat.xls [cv分析统计结果]
│ ├── component_proportions.xls [模型组分统计结果]
│ ├── dif [差异基因与差异代谢物o2pls模型]
│ │ ├── dif.component_proportions.xls [模型组分统计]
│ │ ├── dif.cv.stat.xls [cv分析统计]
│ │ ├── o2m_loading.{pdf,png} [模型loading图]
│ │ ├── top25_loading_element.{pdf,png} [loading值前25的关联图]
│ │ ├── dif.Metabiolite_loading.xls [代谢组 loading值表]
│ │ └── dif.Transcript_loading.xls [转录组 loading值表]
│ └── Z-vs-ZJ [Z与ZJ差异基因与差异代谢物o2pls模型]
│ ├── Z-vs-ZJ.component_proportions.xls [cv分析统计]
│ ├── Z-vs-ZJ.cv.stat.xls [模型组分统计]
│ ├── o2m_loading.{pdf,png} [模型loading图]
│ ├── top25_loading_element.{pdf,png} [loading值前25的关联图]
│ ├── Z-vs-ZJ.Metabiolite_loading.xls [代谢组 loading值表]
│ └── Z-vs-ZJ.Transcript_loading.xls [转录组 loading值表]
├── 04.correlation [peason相关系数模型]
│ ├── 1.dif_pearson [差异基因与差异代谢物相关性结果]
│ │ ├── dif.pearson.xls [peason相关性结果]
│ │ ├── dif.top250.heatmap.{pdf,png} [相关性前250基因与代谢物热图]
│ │ ├── dif.top250.corMatrix.xls [相关性前250基因与代谢物矩阵]
│ │ ├── top250.{Node,Edge}4cytoscape.tsv [相关性前250网络图绘图数据]
│ │ ├── pearson.sigma_net.html [相关系数网路图]
│ │ └── pearson.sigma_net.png [相关系数网路图]
│ └── 2.dif_all_pearson [差异基因与所有代谢物相关性结果]
│ ├── dif_all.pearson.xls [peason相关性结果]
│ ├── dif_all.top250.heatmap.{pdf,png} [相关性前250基因与代谢物热图]
│ ├── dif_all.top250.corMatrix.xls [相关性前250基因与代谢物矩阵]
│ ├── top250.{Node,Edge}4cytoscape.tsv [相关性前250网络图绘图数据]
│ ├── pearson.sigma_net.html [相关系数网路图]
│ └── pearson.sigma_net.png [相关系数网路图]
├── index.html [html版结题报告索引]
└── src [结题报告配置文件夹]
├── content.html [结题报告主体html文件]
├── css [css配置文件夹]
├── doc [说明文档配置文件夹]
├── image [图片配置文件夹]
└── js [js配置文件夹]
7 附录
7.1 参考文献
[1] Cho K, Cho K, Sohn H et al. 2016. Network analysis of the metabolome and transcriptome reveals novel regulation of potato pigmentation[J]. Journal of Experimental Botany, 67(5): 1519-1533.
[2] Zhang X, Zhou Q, Zou W et al. 2017. Molecular Mechanisms of Developmental Toxicity Induced by Graphene Oxide at Predicted Environmental Concentrations[J]. Environ Sci Technol, 51(14): 7861-7871.
[3] Li Q, Guo S, Jiang X et al. 2016. Mice carrying a human GLUD2 gene recapitulate aspects of human transcriptome and metabolome development[J]. Proceedings of the National Academy of Sciences, 113(19): 5358-5363.
[4] El Bouhaddani, Said, et al. "Evaluation of O2PLS in Omics data integration." BMC bioinformatics. Vol. 17. No. 2. BioMed Central, 2016.
[5] Szymanski, Jedrzej, et al. "Linking gene expression and membrane lipid composition of Arabidopsis." The Plant Cell26.3 (2014): 915-928.
[6] Hamanishi E T, Barchet G L, Dauwe R et al. 2015. Poplar trees reconfigure the transcriptome and metabolome in response to drought in a genotype- and time-of-day-dependent manner[J]. BMC Genomics, 16(1).
[7] Bartel J, Krumsiek J, Schramm K et al. 2015. The Human Blood Metabolome-Transcriptome Interface[J]. PLOS Genetics, 11(6): e1005274.
[8] Copley T R, Aliferis K A, Kliebenstein D J et al. 2017. An integrated RNAseq-1H NMR metabolomics approach to understand soybean primary metabolism regulation in response to Rhizoctonia foliar blight disease[J]. BMC Plant Biology, 17(1).
7.2 帮助文档
代谢组与转录组关联分析帮助文档:help.html
代谢组与转录组关联分析英文方法:method.pdf
7.3 引用与致谢
如果您的研究课题使用了基迪奥的测序和分析服务,我们期望您在论文发表时,在Method部分或Acknowledgements部分引用或提及基迪奥公司。
以下语句可供参考:
We are grateful to/thank Guangzhou Genedenovo Biotechnology Co., Ltd for assisting in sequencing and/or bioinformatics analysis.
