1. FASTP数据质控(Reads 过滤)
去除N比例过高的序列:去除Reads中N碱基占比超过10%的序列;
去除低质量序列:去除质量值高于20的碱基数占碱基总数的百分比小于50%的Reads。
碱基质量值与正率的关系下:
| 碱基正确率 | Q-sorce |
|---|---|
| 90% | Q10 |
| 99% | Q20 |
| 99.90% | Q30 |
| 99.99% | Q40 |
2. Tags拼接
根据PE reads之间的重叠关系,使用FLASH(v1.2.11)将成对双端reads拼接为一条序列。拼接的条件是最小匹配长度为10bp,重叠区域允许的错配率为 2%。拼接得到的序列称为Raw Tags。
3. Tags过滤
拼接得到的Raw Tags,经过质量、长度过滤处理后,得到高质量的Tags数据(Clean Tags)。参照Qiime(v1.9.1)的Tags质量控制流程,进行如下操作:
Tags截取:将Raw Tags从连续低质量值(默认质量阈值为<=3)碱基数达到设定长度(默认长度值为3)的第一个低质量碱基位点截断;
Tags长度过滤:经过截取后得到的Tags数据集,进一步过滤其中连续高质量碱基长度小于Tags长度75%的Tags,过滤长度低于300bp的tag。
4. OTU聚类
OTU:Operational Taxonomic Units,操作分类单元。基于 tag(核酸序列)按序列相似度聚类,分成不同的序列集合(cluster),一个cluster即为1个OTU,并挑选cluster中丰度最高的序列作为该OTU的代表序列。进行聚类操作既可以提高运算效率,还可以减小测序错误的影响(相对于高丰度序列,低丰度序列出现测序错误的概率更高,cluster中选取高丰度序列作为代表,避免了低丰度序列直接参与后续分析)。
使用Usearch软件中的Uparse算法,对所有样品的全部Clean Tags 序列聚类, 默认以97%的一致性(Identity)将序列聚类成为OTU。
5. Tags去嵌合体
嵌合体是指PCR的序列扩增过程中,由于DNA缠绕等原因,导致一个扩增子的上下游来源于不同模板的不正常现象,即产生嵌合体。我们基于USEARCH软件的UCHIME算法,在OTU聚类的Tag比对过程中,如果一条tag的上下游同时比对上多条序列,则检测为嵌合体tags,被过滤掉。去除嵌合体以后的高质量Tag即为Effective tag。基于Effective tag统计OTU的丰度信息。
| 表头属性 | 说明 |
|---|---|
| Sample Name | 样本名称 |
| Raw PE | 原始的Pair End Reads对数 |
| Clean PE | 经过质控过滤后的得到的高质量的Pair-end Reads对数 |
| Raw Tags | 经过overlap组装后的得到的原始Tags数量 |
| Clean Tags | 经过Tags质控后的到的高质量的Tags数量 |
| Chimera OTU | 聚类过程中检测到的嵌合体Tags数量 |
| Effective Tags | 去除嵌合体以后的高质量Tags数,即用于后续分析的有效Tags |
| Effective Ratio (%) | 高质量Tags数量占据原始PE Reads的百分比 |
Reads QC filter,低质量reads;
Non-overlap, 没有overlap的未组装reads;
Tag QC filter,未通过“tag过滤”的tags;
Chimera,嵌合体的tag数
Effective tag,有效数据的tags数
| 表头属性 | 说明 |
|---|---|
| SampleID | 样品的id |
| Tags Number | 该样品获得的Effective Tags数 |
| Total length | Tags的总长 |
| Max length | 最长Tag的长度 |
| Min length | 最短Tag的长度 |
| N50 | 该样品Tags序列的N50长度 |
| N90 | 该样品Tags序列的N90长度 |
| 表头属性 | 说明 |
|---|---|
| SampleID | 样品名称,最后一行avg表示所有样本均值。 |
| Total Tags | effective tags的数量 |
| Unique Tags | unique tags的数量(100%相似度获得的OTU,也称为Unigue tag) |
| Taxon Tags | 有物种注释的tags的数量 |
| Unclassified Tags | 没有物种注释的tags的数量 |
| Singleton Tags | 总丰度为1的OTUs所对应的Tags的数量 (被过滤掉的OTUs) |
| OTUs | 最终得到的OTUs的数量 |
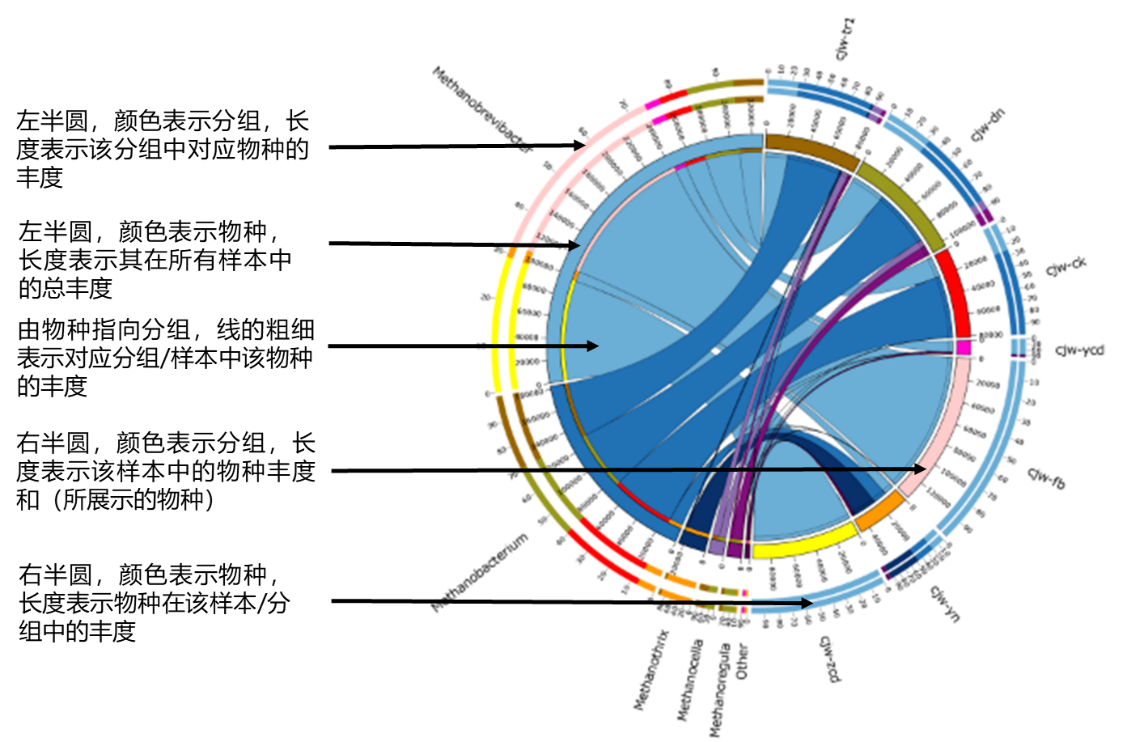
注:图中左边半圆表示物种,右边半圆表示样本/分组。从物种到分组连线的宽度,表示分组/样本中物种的相对丰度。左边半圆的外环,颜色表示分组,环的长度表示每个物种在各个分组丰度,右边半圆的外环,颜色表示物种,环的长度表示每个分组中各个物种的丰度。
相关性网络图,点的大小表示物种丰度,红色实线表示正相关,蓝色虚线表示负相关。
左边韦恩图,圆表示不同分组,圆之间重叠部分表示共有物种或基因数量,非重叠部分表示特有物种或基因的数量。共有、特有部分物种、基因信息见文件夹中。
物种丰度分布差异检验方法对比:
卡方检验:两样本比较,物种在两样本间是否有显著差异
welch’s T检验:两组比较,物种在两组间的均值是否有显著差异
Wilcoxon秩和检验:两组比较,物种在两组间的中位数是否有显著差异
Kruskal-Wallis秩和检验:多组比较,判断各分组的中位数是否有显著差异
Tukey HSD:多组比较中的验后检验(两两比较),首先基于方差分析(ANOVA)判断多组的中位数是否有显著差异,对方差分析显示有显著差异的物种,再使用Tukey HSD进行多组间的两两比较,判断均值是否有显著差异。
左边柱状图展示了差异物种在两个分组中的相对丰度;右边点的坐标为丰度差值,误差棒展示差值在95%置信区间中的波动范围,最右侧标注T检验结果的P值大小,其中P值越小说明差异越显著。
随机森林是一种基于决策树的高效的机器学习算法,属于非线性分类器,可以挖掘变量之间复杂的非线性的相互依赖关系。通过随机森林分析,可以找出能够区分两组样本间差异的关键成分(物种)。针对比较组获得的显著差异物种,使用R语言randomForest包进行随机森林分析。
纵轴表示物种,横轴表示指标的分值。“mean decrease accuracy”主要思路是通过顺序变动来度量每个物种对模型精确率降低程度的影响。不重要的物种对模型精确率的影响不会太大,但是对于重要的物种,会降低模型的精确率,所以该值越大表示该物种的重要性越大;“mean decrease gini”,度量每个物种对分类树每个节点上观测值的异质性的影响,值越高,说明该物种的重要性越大。
横轴表示物种区分能力的准确度,纵轴表示区分能力的灵敏度。随着准确度增高,灵敏度会降低。ROC曲线下的面积值(AUC)在1.0和0.5之间。AUC>0.5的情况下,AUC越接近于1,说明区分效果越好。AUC在0.5~0.7时区分能力较低,AUC在0.7~0.9时有一定区分能力,AUC在0.9以上时有较高区分能力。AUC=0.5时,说明完全无区分效果。
气泡图纵轴表示物种,横坐标表示分组,气泡的大小表示每个物种在分组中的 indicator 值大小,即该物种在该分组中的指示性大小。气泡颜色表示分组。
多样性指数计算情参考基迪奥论坛文章:一文详解α多样性
横坐标是OTU相对丰度从大到小的排序;纵坐标表示该OTU的相对丰度。
横坐标是分组;纵坐标是多样性指数大小。图中点表示样本。字母相同表示有显著差异,不同表示无显著差异。
基于OTU丰度表进行4种距离计算。基于各物种分类层级,进行Jaccard和Bray距离计算。
距离指数参考基迪奥论坛文章:微生物β多样性常用计算方法比较
计算方法:
Jaccard、Bray:使用R语言Vegan包。
(un)weighted unifrac:先使用Muscle软件进行OTU代表序列比对,然后将比对文件导入FastTree软件构建系统发育树,再使用R语言GuniFrac包进行(un)weighted unifrac距离分析。
PC1、PC2坐标表示第一、第二主成分,括号中的百分比则表示主成分对样品差异的贡献值。图中有颜色的点分别表示各个样品。一般两个维度的贡献值之和达到50%比较好。
Diffs:差异比较组
Df:自由度
SumsOfSqs:总方差,又称离差平方和
MeanSqs :均方(差),即Sums Of Sqs/Df
Fvalue:F 检验值
R2:不同分组对样品差异的解释度,即分组方差与总方差的比值,R2 越大表示分组对差异的解释度越高
Pvalue:P值,数值小于0.05 说明本次检验的可信度高
Diffs:差异比较组
R:差异程度,一般介于(0,1)之间
Pvalue:P值,数值小于0.05 说明本次检验的可信度高
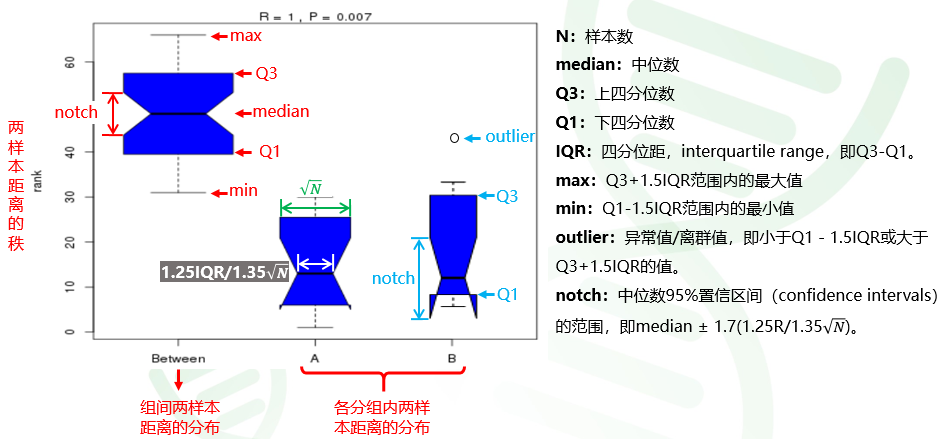
R值表示组间与组内距离的差异程度,数值越大表示差异越大。P值表示组间与组内距离差异的显著性。以带notch(凹槽)的盒型图详细展示组内、组间距离的分布特征,图形含义如下图。
Bugbase基于已知物种基因组的表型特征,构建物种-基因-表型的网络,基于OTU Greengene的物种注释信息预测每个样本中不同表型的丰度,以及物种对每种表型的贡献度。
07.Function/BugBase/thresholds文件夹中的图形(如下图)。
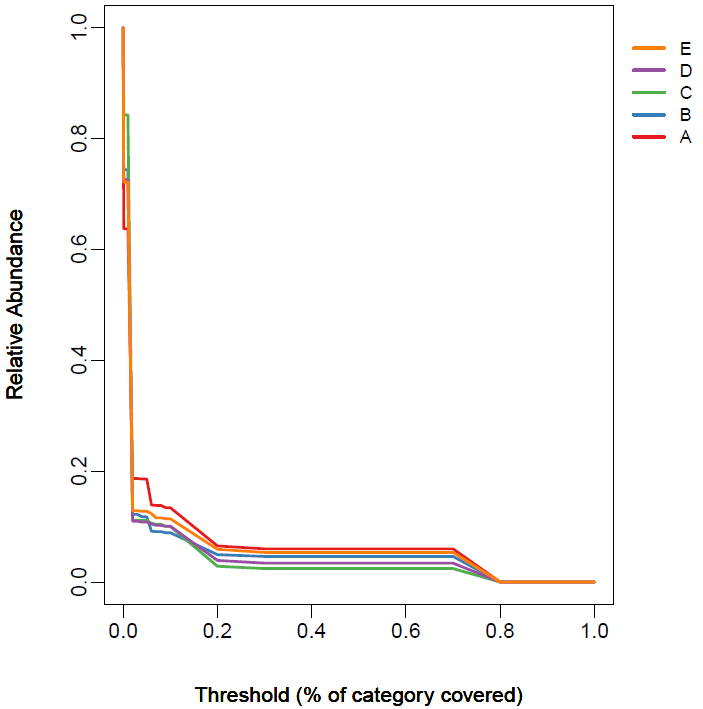
线颜色表示样本;横轴表示阈值,即判断为该表型的最少基因数占比,取值0到1;纵轴表示取不同阈值时,对应计算得到的表型丰度。比如bugbase里有一种表型,软件汇总的属于该表型的基因一共有100个,当阈值设为0.8,表示样本里如果包含其中80个基因,则认为样本中存在该表型,由此计算得到对应阈值下,不同样本的表型丰度。
所以当阈值由左向右移动时,代表判断要求越来越高,所以对应丰度呈下降趋势。按软件默认参数选择每种表型的最佳阈值,存放在文档threshold_used.txt中。
堆叠图横坐标为不同的样品分组,不同颜色的柱子表示了不同物种对同一个表型贡献的OTU相对丰度。由于注释层级越高OTU数目越少,选择门水平既保留大部分的丰度,也能展示同一个表型所关联的物种类型及其物种丰度。
表格说明
第一列:样品名称
第二列到最后一列:各表型分类相对丰度
表格说明
第一列:比较组方案
第二列到最后一列:差异检验的P值
箱线图横坐标为不同的样品分组,对应的点为分组中样品的丰度高低,高中低三条线分别表示组内样品丰度的上四分位数,中位数和下四分位数。
通过比较不同分组的箱线位置高低,可以比较不同样品分组在不同表型中丰度的高低情况。
func:生态功能名称
第二列到最后一列:不同样品的功能丰度
堆叠图横坐标为不同的样品,不同颜色的柱子表示不同的生态功能的相对丰度,其中我们展示丰度前11的功能丰度,其他功能的丰度合并到other分类中。