基迪奥生物 scRNA-seq分析 帮助文档
项目流程
实验流程
1)10x分离单细胞
Chromium™ Single Cell 3'Solution是建立在GemCode技术上的微流体平台,将带有条形码和引物的凝胶珠与单个细胞包裹在油滴中,形成GEM(Gel bead in emulsion),GEM是指油滴包裹的,含有Barcode的Gel beads和细胞、反应试剂的混合物。回收GEM并纯化,用于后续实验。
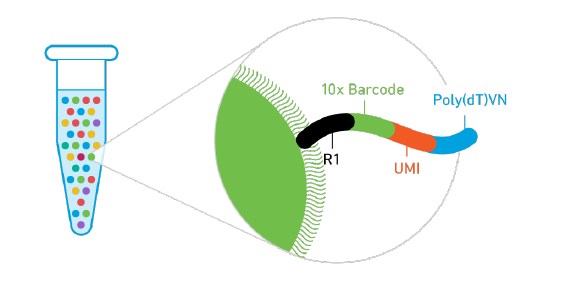
图1 GEM结构
2)cDNA形成与扩增反应
GEM内的凝胶珠发生溶解,细胞裂解释放mRNA,通过逆转录产生用于测序的带条形码的cDNA;液体油层破坏后,进行cDNA扩增反应,纯化后进行质检,质检合格后进行文库构建。
3)文库构建
酶切打断,片段进行末端补平,并加碱基A,连接接头,试剂盒筛选回收目标大小片段,进行文库质检与定量。
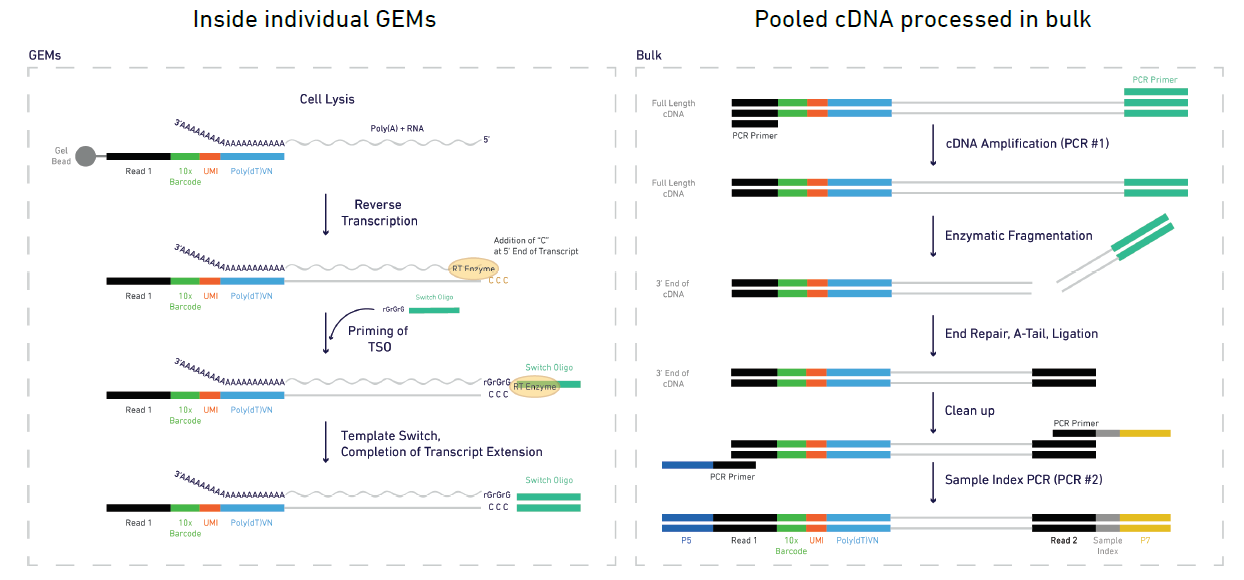
图2 文库构建
4)文库测序
构建完成的文库如下图所示,有左到右(5'~3')分别是:Illumina P5接头、P5测序引物(黑色Read1处)、16bp 10x™Barcode、10bp UMI、ploy(dT)VN序列、插入cDNA片段、P7测序引物(黑色Read2处)、样本Index(i7 index read)、Illumina P7接头。Read 1的有用信息只有前26bp(16bp Barcode + 10bp UMI),用于确定此文库序列来源的细胞与RNA分子;Read 2的有用信息只有前98bp cDNA序列,用于比对参考基因组以确定本分子所属基因。
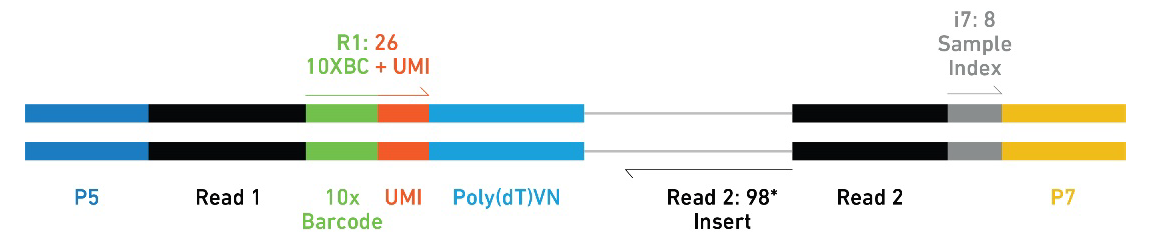
图3 文库结构
分析流程
1)利用10x Genomics官方分析软件Cell Ranger(https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome)对原始数据进行数据过滤、比对、定量、鉴定回收细胞,最终得到各细胞的基因表达矩阵。
2)后续采用Seurat(https://satijalab.org/seurat/)进行进一步的细胞过滤、标准化、细胞亚群分类、各亚群差异表达基因分析及标记基因筛选。
测序数据质控和表达量定量
测序数据基本质控
各样本测序数据统计表
| 表头属性 | 说明 |
|---|---|
| Sample | 样本名称 |
| Number of reads | 样本测序得到的reads数量 |
| Valid Barcodes | 带有正确barcode的reads百分比 |
| Sequencing Saturation | 预估的测序饱和度 |
| Q30 Bases in Barcodes | Barcode序列中质量分数大于等于 30 的碱基百分比 |
| Q30 Bases in RNA Read | 用于比对的Reads中质量分数大于等于 30 的碱基百分比 |
| Q30 Bases in UMI | UMI 序列中质量分数大于等于 30 的碱基百分比 |
数据定量
我们采用10x Genomics官方分析软件Cell Ranger对原始数据进行数据质量统计,并比对参考基因组。
Illumina双末端测序结果中,Read1包含16bp GemCode barcode(用于区分不同细胞)和10bp UMI(Unique Molecular Identifier,用于区分不同RNA分子,一个mRNA分子将会被一个UMI标记);Read2为cDNA序列片段。
Cell Ranger调用STAR(Spliced Transcripts Alignment to a Reference)比对软件将Read2比对到参考基因组。依据GTF注释的结果,将比对上的reads分类为外显子、内含子或基因间区的reads:如一条reads有至少50%位于外显子区则属于外显子reads,以此类推。如果reads比对到单一的外显子区域和一个/多个非外显子区,则优先归类为外显子且它的MAPQ被置为255。Cell Ranger进一步将外显子reads比对回已注释的转录本上:1. 如果reads比对到某条转录本的外显子且两者方向相同,则认为它比对到了转录组上,记为转录组reads; 2. 在转录组reads中,如果reads只比对到一个基因,则认为它们是uni-mapped的。只有uni-mapped的reads才用于UMI couting。
Cell Ranger接着会过滤和校正barcodes与UMIs。Cell barcodes要求与数据库已知的barcode序列完全一致,只允许有一个错配且这个错配只能出现在低质量碱基处。接着这个错误将会被校正,而其他不满足该条件的barcodes将会被过滤。UMI不允许是单寡聚链、不允许含有N、不允许含有质量值低于<10的碱基,否则会被过滤。如果某个UMI与更高计数的UMI只有一个错配且它们有相同的barcode和gene id,则它会被校正成较高计数的那个UMI。只有有效验证过barcode和UMI的reads才用于UMI couting。
将每个barcode的每个gene id对应的UMI去重,计算unique UMI的数量作为该细胞该基因的表达量。
记预期得到的细胞数为N。将barcode按UMI count数从大到小排列,取前N个barcode。记其第99百分位数(percentile)的UMI count数为m,过滤UMI count < 10% * m的barcode,剩下的则为有效的细胞(图1 绿线)。
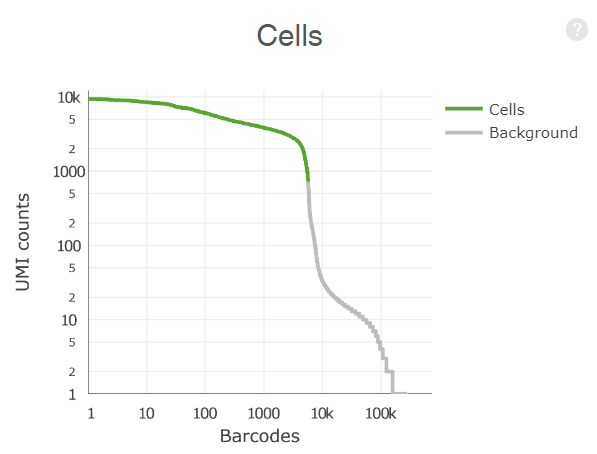
图4 鉴定回收细胞
估计计算示例:本次预设回收数量为8000细胞,8000 * 1% barcode对应的UMI数约为6500,10% * 6500 UMI对应barcode约为6000。本次软件实际回收5705细胞。
10x Genomics关于Cell Ranger软件算法的说明: https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/algorithms/overview
各样本比对结果统计表
| 表头属性 | 说明 |
|---|---|
| Sample | 样本名称 |
| Estimated Number of Cells | 高质量细胞数,小于所有细胞的 UMI 总数的 99% 分位数的 10% 定义为背景噪音细胞 |
| Fraction Reads in Cells | 高质量细胞所含reads所占整体reads的百分比 |
| Mean Reads Per Cells | 每个细胞的平均reads数 |
| Median Genes Per Cells | 各个细胞检测到的基因数的中位数 |
| Total Genes Detected | 所有细胞检测到的基因总数 |
| Median UMI Counts Per Cells | 各个细胞检测到的UMI数的中位数 |
| Reads Mapped Confidently to Genome | 比对到参考基因组的reads百分比 |
| Reads Mapped Confidently to Integenic Regions | 比对到基因间区的reads百分比 |
| Reads Mapped Confidently to Intronic Regions | 比对到内含子区域的reads百分比 |
| Reads Mapped Confidently to Exonic Regions | 比对到外显子区域的reads百分比 |
| Reads Mapped Confidently to Transcriptome | 比对到已知参考转录本的reads百分比 |
最终鉴定细胞表达量矩阵
UMI丰度信息表
| 表头属性 | 说明 |
|---|---|
| Gene ID | 基因ID |
| Name | 基因的名称(symbol号) |
| 第3列~其后所有列 | 表头为各个细胞对应的标签序列,对应内容为细胞中各个基因的UMI丰度 |
细胞质控信息可视化
单细胞亚群分类
非正常细胞的进一步过滤
多胞检测结果统计表
| 表头属性 | 说明 |
|---|---|
| Cells | 细胞barcode |
| pANN | 细胞为多胞的概率 |
| Classification | Singlet(单胞),Doublet(多胞) |
过滤前后各个样本中细胞数据量统计表
| 表头属性 | 说明 |
|---|---|
| Sample | 样本名称 |
| before_filter_num | 过滤前细胞数量 |
| after_filter_num | 过滤后细胞数量 |
| pct | 过滤后数量百分比 |
| after_filter_median_UMI_per_cell | 过滤后每个细胞UMI数量的中位数 |
| after_filter_median_genes_per_cell | 过滤后每个细胞Gene数量的中位数 |
单细胞亚群分类
分析过程:
- 表达量均一化
在去除低质量细胞后,我们首先进行数据的表达量均一化。我们使用Seurat软件的Normalization()函数的LogNormalize方法,进行表达量均一化。
表达量均一化计算公式:A = log( 1 + ( UMIA ÷ UMITotal ) × 10000 )
其中,A:目标细胞中目标基因A的表达量;UMIA: 目标细胞中A基因的UMI数量;UMITotal:目标细胞中所有UMI数量的总和;log:以e为底数的自然对数。 - 高变基因筛选
我们使用Seurat软件FindVariableFeatures()函数的vst方法寻找细胞间高特征基因,这些基因将用于下游分析,可以有效降低背景噪音,提高细胞分群精度。 - 批次效应矫正
为了最小化批次处理效应对聚类的影响,我们使用了Harmony算法将细胞投射到共享嵌入中,从而聚合所有样本。 - 表达量归一化
我们使用Seurat软件ScaleData()函数对数据进行Z-score归一化,使基因在各个细胞间的表达量均值等于0,方差等于1,排除部分高表达的基因对下游分析产生主导作用。 - 通过主成分分析降维,减少变量
然后利用均一化后的表达量值进行PCA(Principal component analysis)分析。 - 聚类和分群分析
Seurat软件使用基于图论的聚类算法对细胞进行聚类和分群。主要包括以下步骤:- 构建细胞间的聚类关系:利用显著的主成分构建基于欧式距离的KNN聚类关系图;
- 优化细胞间聚类关系距离的权重值:利用Jaccard相似性优化细胞间距离的权重值;
- 聚类和分群:使用Louvain 算法进行细胞群聚类优化。
Seurat教程及算法说明:https://satijalab.org/seurat/v3.1/pbmc3k_tutorial.html
细胞亚群分类结果统计表
| 表头属性 | 说明 |
|---|---|
| Clusters | 单细胞亚群 |
| Cells number | 各细胞亚群中的细胞数 |
| Median Genes per Cells | 各个细胞亚群中,每个细胞检测到的基因数量的中位数 |
| Median UMI Counts per Cells | 各个细胞亚群中,每个细胞检测到的UMI数量的中位数 |
各样本/组在各个亚群中细胞数量统计表
| 表头属性 | 说明 |
|---|---|
| Cluster | 单细胞亚群 |
| 第2列~其后所有列 | 各样本/组在对应亚群中的细胞数量 |
基因在各个亚群中表达量的均值表
| 表头属性 | 说明 |
|---|---|
| Gene_ID | 基因ID |
| Gene_name | 基因名称 |
| 第3列~其后所有列 | 各亚群表达量均值 |
基因在不同细胞亚群的表达量均值通过基因在不同细胞的表达量TPM值计算得到, TPM值计算公式为:
TPMA = UMIA ÷ UMITotal × 10000
其中,TPMA为目标细胞中基因A的TPM值,UMIA为目标细胞中基因A的UMI数,UMITotal为目标细胞中有效UMI数量的总和
细胞与亚群对照表
| 表头属性 | 说明 |
|---|---|
| Cells | 细胞名称(标签序列) |
| Cluster | 细胞所属的亚群 |
| Samples | 细胞来源的样本 |
| Groups | 细胞所属的组名 |
分类结果可视化
基于已知标记基因的分类效果评估
单细胞亚群鉴定
各样本在各个细胞类型中细胞数量统计表
| 表头属性 | 说明 |
|---|---|
| Cell type | 细胞类型 |
| 其后各列 | 各样本在对应细胞类型中的细胞数量,括号内该样本中对应细胞类型占所有细胞的百分比比例 |
各细胞亚群中各个细胞类型数量统计表
| 表头属性 | 说明 |
|---|---|
| Cluster | 单细胞亚群 |
| Cell.annotation | 细胞亚群对应的细胞类型;括号内为该细胞亚群中,该细胞类型所占百分比比例 |
| 其后各列 | 各细胞亚群中鉴定到的各细胞类型的细胞数量;括号内为该细胞亚群中,该细胞类型所占百分比比例。 |
亚群上调表达基因分析
上调表达基因分析
各亚群上调基因数量统计表
| 表头属性 | 说明 |
|---|---|
| Cluster | 单细胞亚群 |
| Up Gene Number | 上调基因数量 |
各亚群差异基因注释表
| 表头属性 | 说明 |
|---|---|
| Target Cluster | 目标亚群,我们将筛选目标亚群上调的基因 |
| Gene ID | 目标亚群上调的基因对应的ID号 |
| Gene name | 基因名称,一般为symbol号 |
| Target_Cluster_mean | 上调的基因在目标细胞亚群的平均表达量 |
| Other_Cluster_mean | 上调的基因在其他细胞亚群的平均表达量 |
| Log2FC | 目标细胞亚群与其他细胞亚群的基因平均表达量的比值,取log2 |
| Pvalue | 差异检验的p-value |
| Qvalue | 差异检验的q-value |
| Description | 该基因的描述信息 |
| KEGG_A_class | KEGG A级分类名称 |
| KEGG_B_class | KEGG B级分类名称 |
| Pathway | KEGG Pathway注释(C级分类) |
| K_ID | 基因对应KEGG蛋白注释ID |
| GO Component | GO功能注释_细胞组分 |
| GO Function | GO功能注释_分子功能 |
| GO Process | GO功能注释_生物学过程 |
基因在不同细胞亚群的表达量均值通过基因在不同细胞的表达量TPM值计算得到, TPM值计算公式为:
TPMA = UMIA ÷ UMITotal × 10000
其中,TPMA为目标细胞中基因A的TPM值,UMIA为目标细胞中基因A的UMI数,UMITotal为目标细胞中有效UMI数量的总和
标记基因蛋白质互作网络分析
String(http://string-db.org )蛋白质相互作用网络数据库,构建标记基因的蛋白互作网络。String数据库是一个搜寻蛋白质之间相互作用的数据库,既包括蛋白质之间的直接物理相互作用,也包括蛋白质之间的间接功能相关性。该数据库可应用于2031个物种,包含960万种蛋白和1380万中蛋白质之间的相互作用。通过Blast比对,寻找标记基因在String数据库中有对应关系的蛋白,将对应蛋白的相关网络关系作为标记基因的相关网络关系。得到标记基因的蛋白互作关系信息后,将结果文件导入Cytoscape,进行网络可视化。
GSVA分析
MSigDB数据库的八个数据集包含的通路信息如下:
- H:共包含50个基因集。该数据集总结了50个定义明确的生物学状态或生物学进程相关的通路信息。[12]
- C1:共包含299个基因集。该数据集的基因集是由Ensembl BioMart中人的基因组注释信息中的不同染色体位置的基因构成的,这些基因集有助于识别与染色体缺失或扩增、剂量补偿、表观遗传沉默和其他区域效应相关的效应。
- C2:共包含5529个基因集。该数据集是由线上数据库和生物医学文献提供的数据整理得到的,包含两个子集——化学和遗传扰动(Chemical and genetic perturbations, CGP)及经典通路(Canonical pathways, CP)——组成。CP子集包含从BioCarta、KEGG[7]、Pathway Interaction Database、Reactome[8]四个线上数据库收集的共计2232个基因集。CGP子集的基因集主要从生物医学文献中收集,描述了受到化学因素扰动而发生表达量变化的3297个基因集,反映了若干重要的生物学特征和临床特征,例如癌症转移、耐药性等。
- C3:共包含3735个基因集。该数据集收集了可以作为潜在调控靶基因的基因,并按照相同的调控元件将靶基因整合为一个基因集。该数据集可以被分为两个部分——microRNA靶基因(microRNA targets, MIR)[13][14][15]和转录因子靶基因(transcription factor targets, TFT)[16][17]。
- C4:共包含858个基因集。该数据集是通过对癌症微阵列数据的大量分析得到的癌症相关基因集。该数据集包含两个部分:(1)癌症基因相关基因集(Cancer gene neighborhoods, CGN),通过4个微阵列数据,选择380个癌症基因,计算其余其他基因的皮尔森相关系数,选择相关系数≥0.85的基因作为一个癌症基因的CGN基因集,排除基因数<25个基因集,最终获得427个基因集;[18](2)癌症模块(Cancer Module, CM),从文章得到的456个与癌症相关的模块,并选择了其中431个作为基因集[19]。
- C5:共包含10192个基因集。该数据集是由GO数据库得到的关于生物学进程(biological process, BP)、细胞组分(cellular component, CC)和分子功能(molecular function, MF)的基因集。[6]
- C6:共包含189个基因集。该数据集代表了在癌症中失调的细胞通路
- C7:共包含4872个基因集。该数据集收集了对免疫系统中细胞类型、细胞状态和免疫扰动相关基因基因集。[20]
转录因子注释
转录因子注释表
| 表头属性 | 说明 |
|---|---|
| gene_id | 基因的ID号 |
| TF_family | 转录因子所属的转录因子家族 |
| Gene_name | 基因的symbol号 |
| Cluster_* | 转录因子在各个细胞亚群的平均表达量 |
| Description | 基因在NCBI数据库的注释信息 |
| KEGG_A_class | 转录因子在KEGG数据库中的A级注释 |
| KEGG_B_class | 转录因子在KEGG数据库中的B级注释 |
| Pathway | 转录因子在KEGG数据库中所属通路名称 |
| K_ID | 转录因子在KEGG数据库中对应的ID号 |
| GO Component | 转录因子在GO数据库中所属细胞组分注释 |
| GO Function | 转录因子在GO数据库中所属分子功能注释 |
| GO Process | 转录因子在GO数据库中所属生物进程注释 |
膜蛋白注释
膜蛋白注释表
| 表头属性 | 说明 |
|---|---|
| gene_id | 基因的ID号 |
| AA_length | 基因编码的氨基酸序列的长度 |
| ExpAA | 预测的跨膜螺旋氨基酸数目,如果数目大于18,则该蛋白很可能是个跨膜蛋白,或者含有信号肽结构 |
| First60 | 蛋白N端前60个氨基酸中预测为跨膜螺旋氨基酸的数目,代表N端的跨膜螺旋为信号肽的可能性 |
| PredHel | 通过N-best预测的跨膜螺旋数量 |
| Topology | 有N-best拓扑预测的最终跨膜结果,“i”表示膜内肽链,“o”表示膜外肽链。例,“i95-117o165-187i”,表示第1-94位氨基酸肽链在膜内,95-117位氨基酸肽链为跨膜螺旋结构,118-164位氨基酸肽链在膜外,165-187位氨基酸肽链为跨膜螺旋结构,188-末位氨基酸肽链在膜内。 |
| Gene_name | 基因的symbol号 |
| Cluster_* | 基因在各个细胞亚群的平均表达量 |
| Description | 基因在NCBI数据库的注释信息 |
| KEGG_A_class | 基因在KEGG数据库中的A级注释 |
| KEGG_B_class | 基因在KEGG数据库中的B级注释 |
| Pathway | 基因在KEGG数据库中所属通路名称 |
| K_ID | 基因在KEGG数据库中对应的ID号 |
| GO Component | 基因在GO数据库中所属细胞组分注释 |
| GO Function | 基因在GO数据库中所属分子功能注释 |
| GO Process | 基因在GO数据库中所属生物进程注释 |
细胞周期分析
细胞周期评估
细胞周期推断
各细胞的细胞周期表
| 表头属性 | 说明 |
|---|---|
| Cells | 细胞名称 |
| Samples | 细胞所属样本名称 |
| G1/S_Score | 细胞对应G1/S期评分 |
| S_Score | 细胞对应S期评分 |
| G2/M_Score | 细胞对应G2/M期评分 |
| M_Score | 细胞对应M期评分 |
| M/G1_Score | 细胞对应M/G1期评分 |
| CellCycle.Score | 细胞对应的各个细胞周期评分的最大值 |
| Phase | 根据评分推测的细胞所处细胞周期 |